

Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy
- PMID: 31316208
- PMCID: PMC6779479
- DOI: 10.1038/s41586-019-1406-x
Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy
Abstract
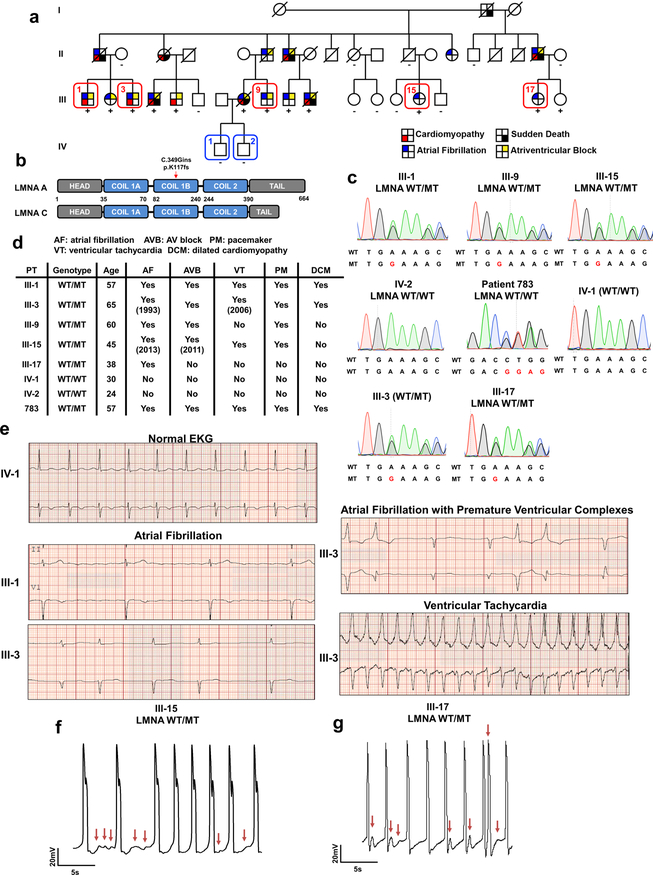
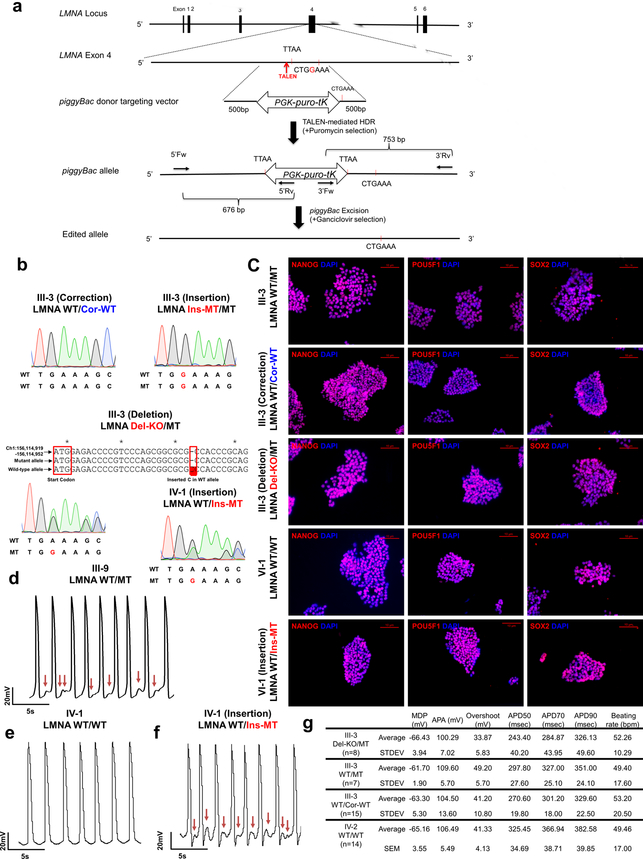
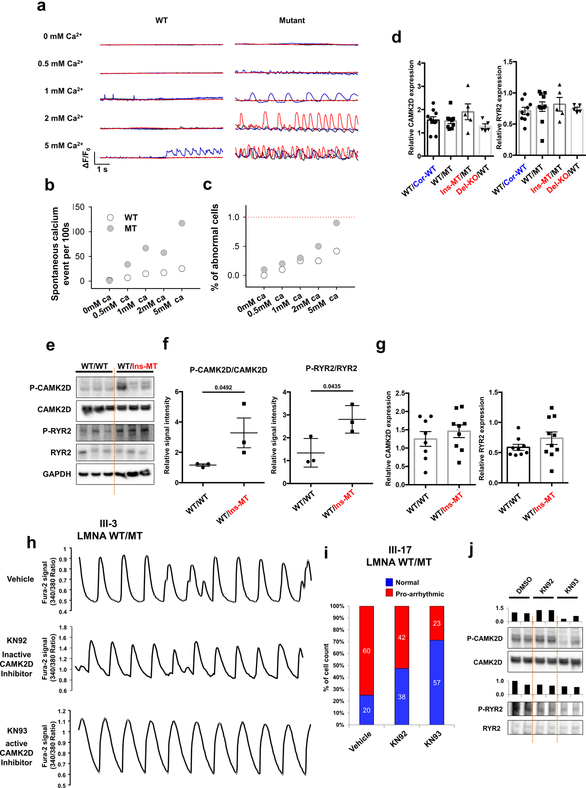
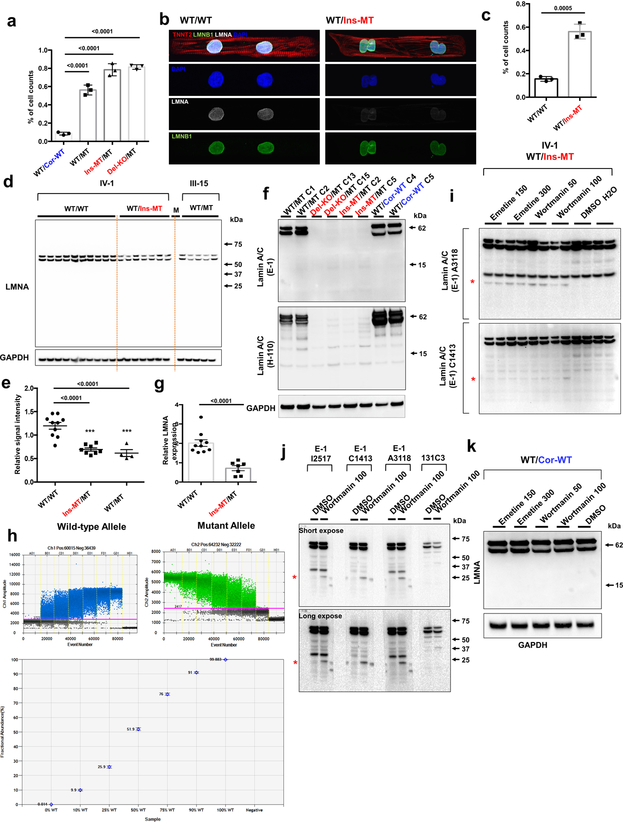
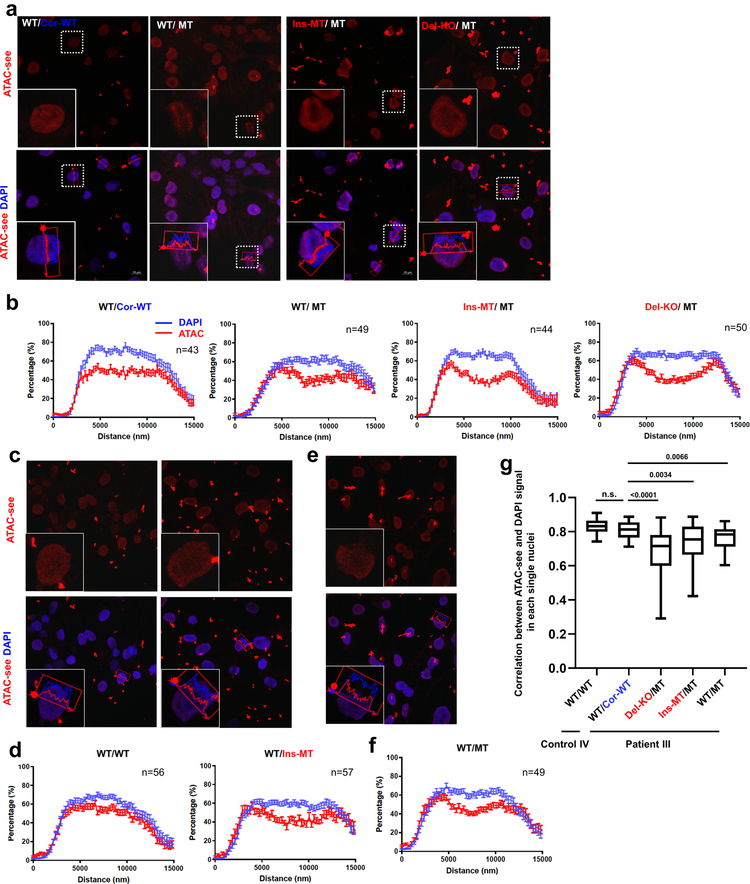
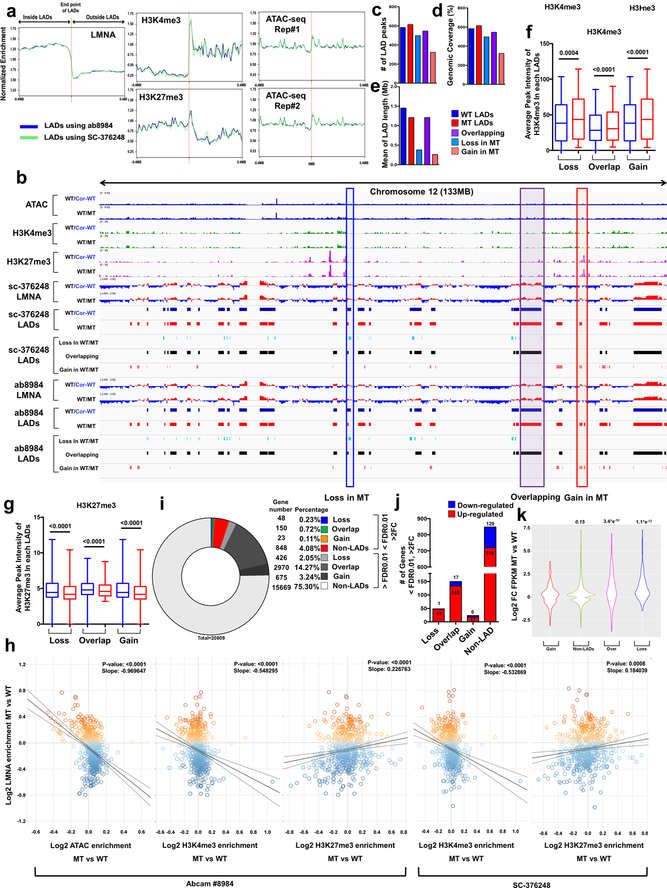
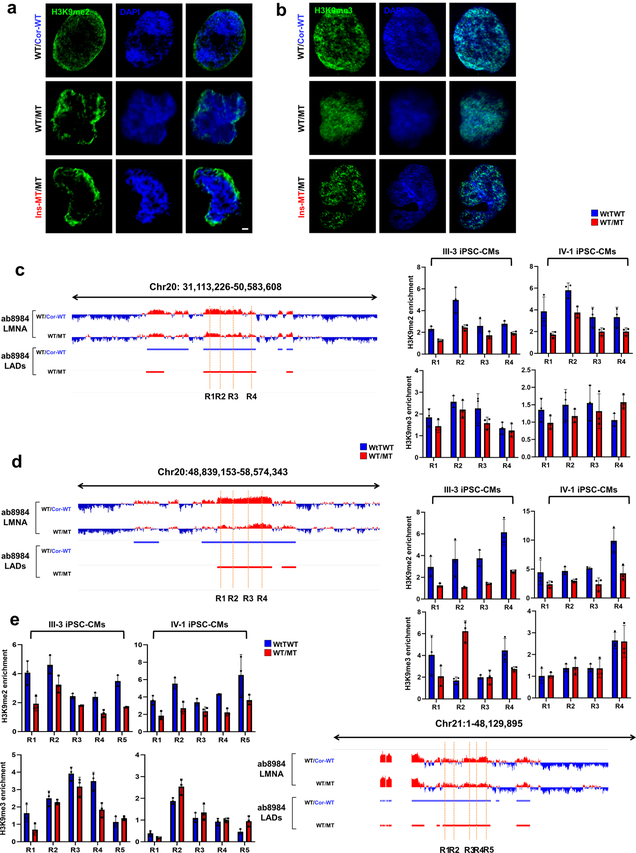
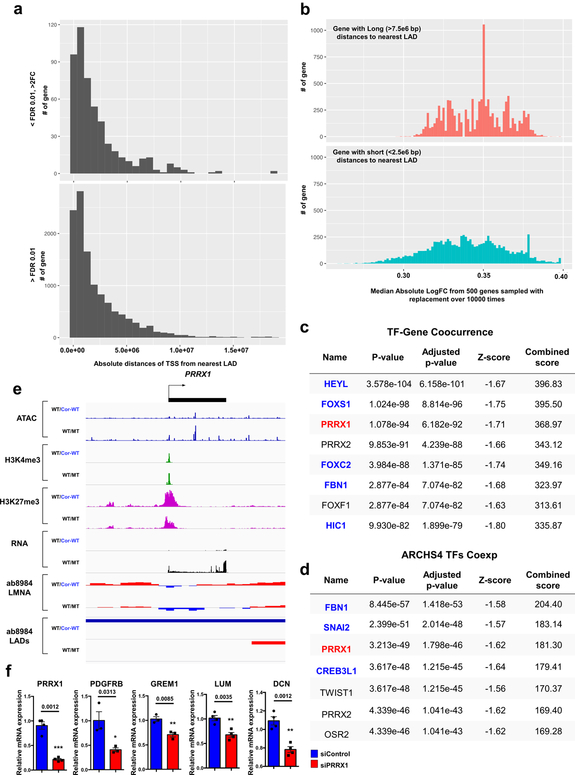
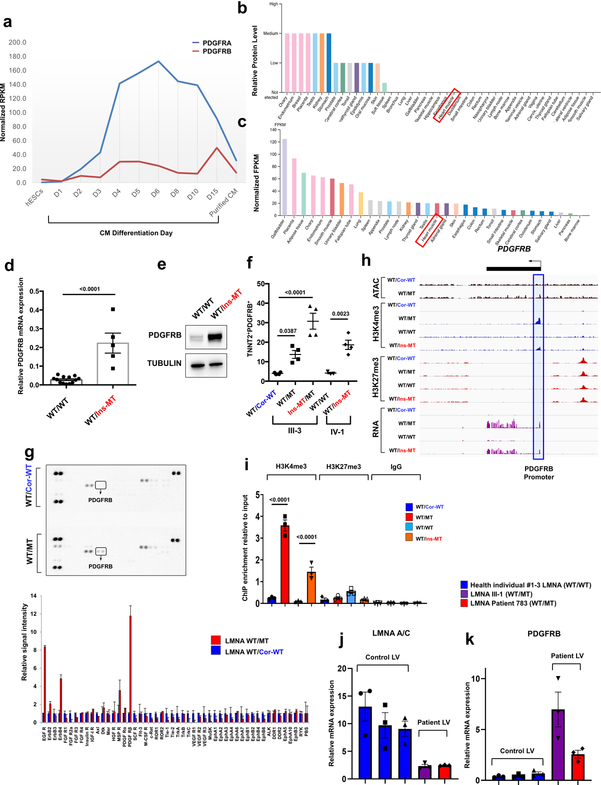
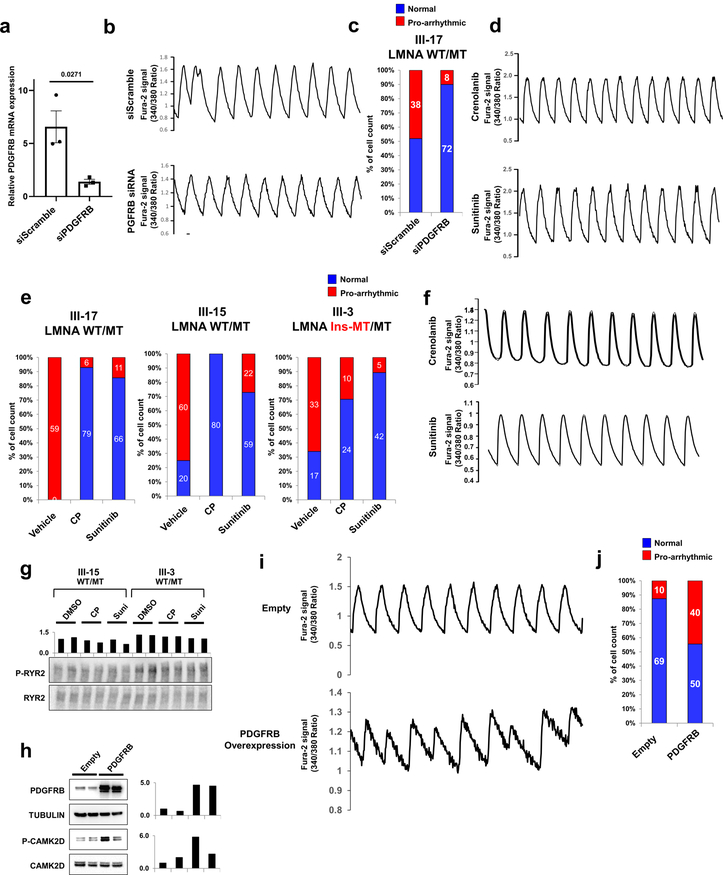
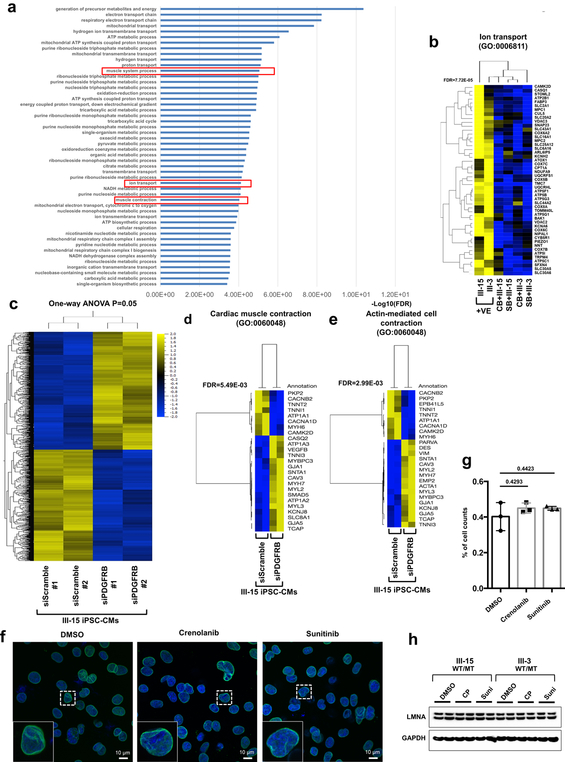
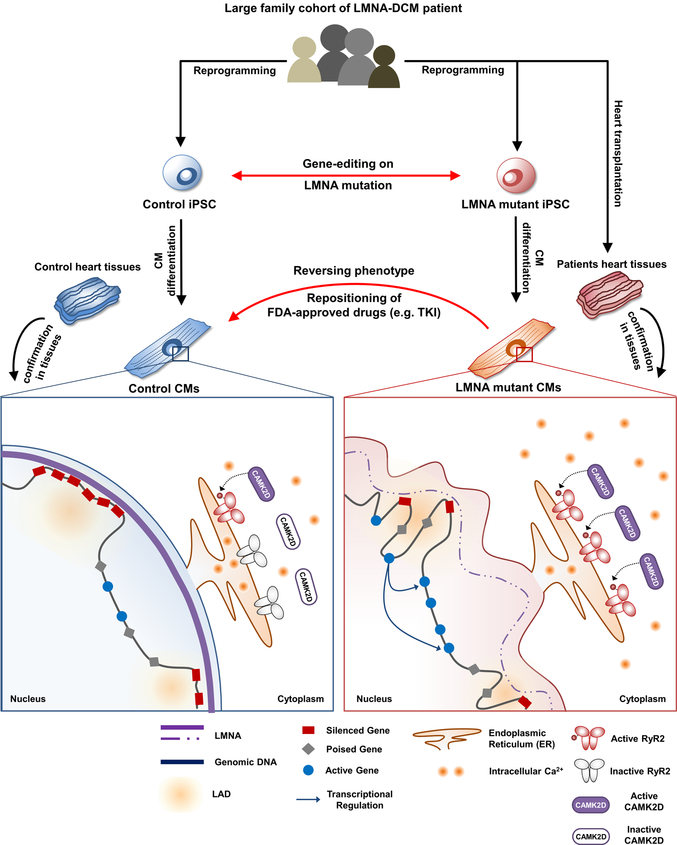
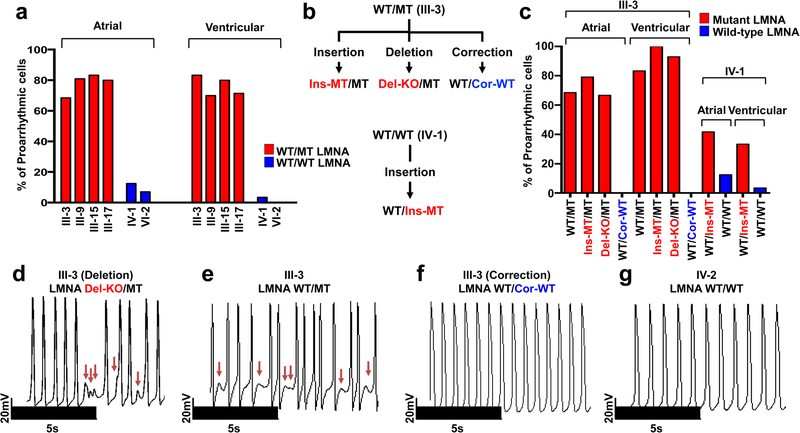
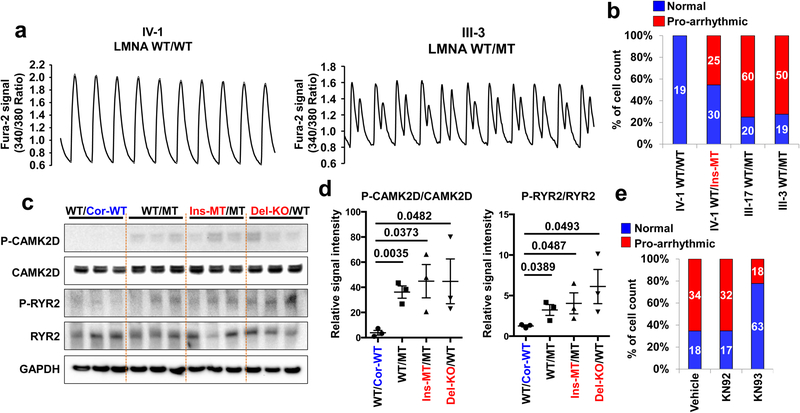
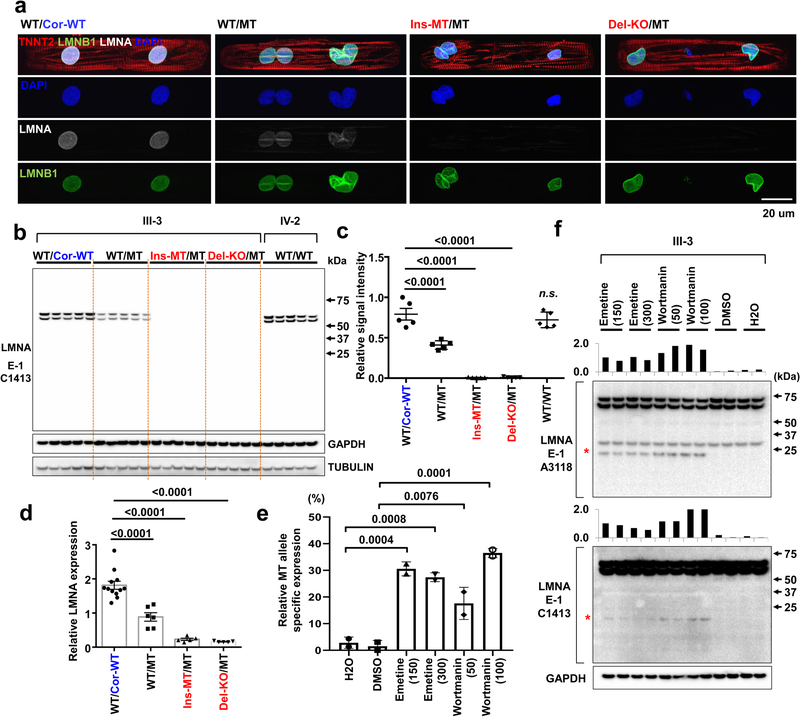
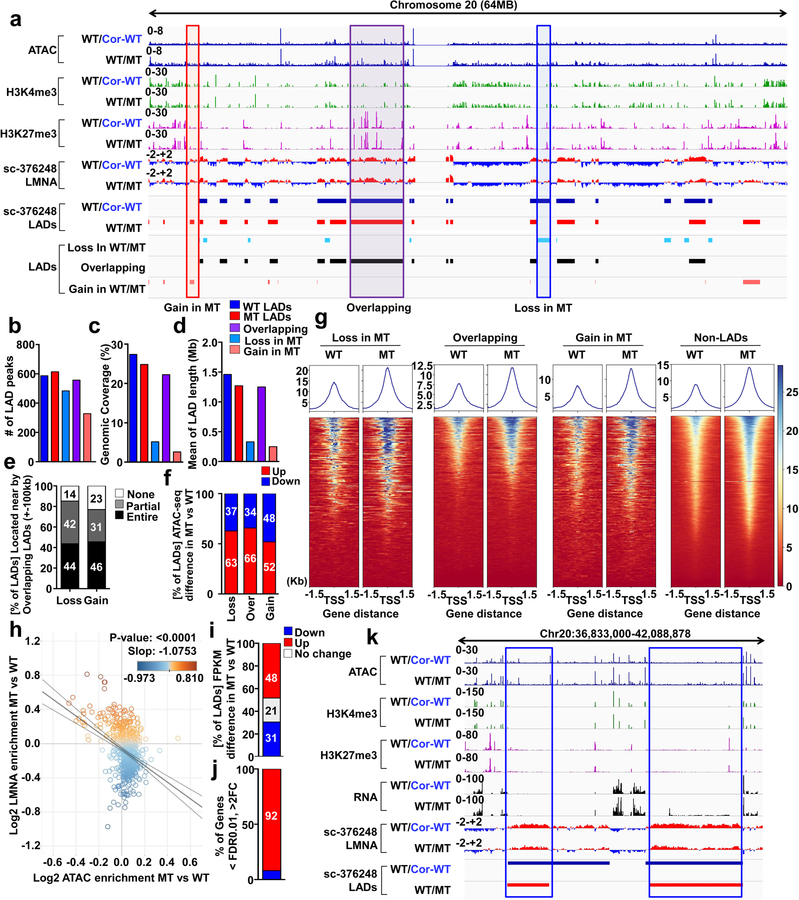
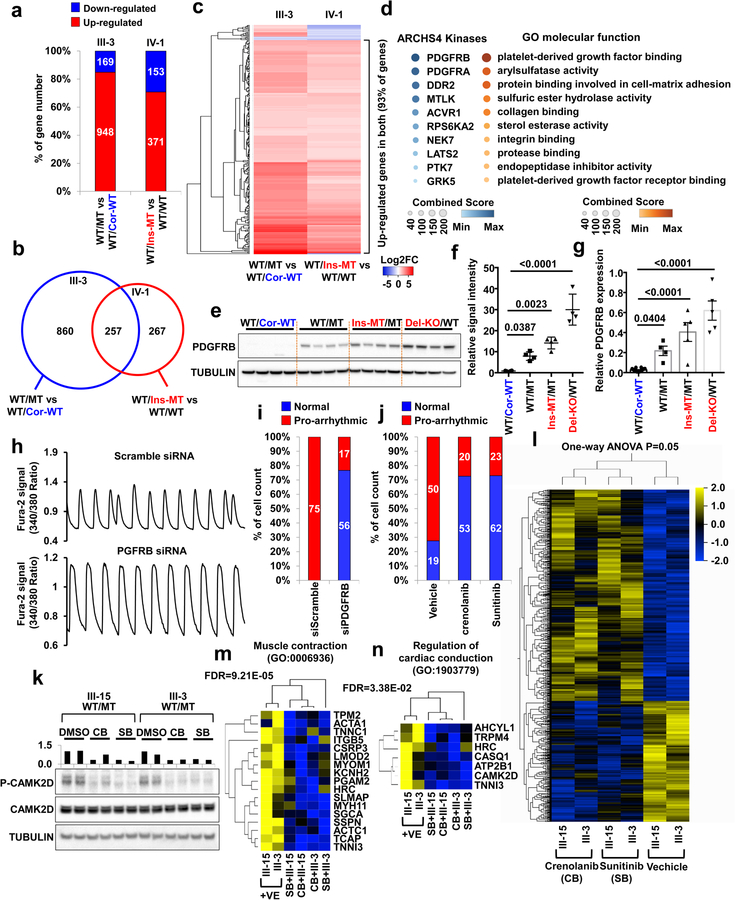
Lamin A/C (LMNA) is one of the most frequently mutated genes associated with dilated cardiomyopathy (DCM). DCM related to mutations in LMNA is a common inherited cardiomyopathy that is associated with systolic dysfunction and cardiac arrhythmias. Here we modelled the LMNA-related DCM in vitro using patient-specific induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs). Electrophysiological studies showed that the mutant iPSC-CMs displayed aberrant calcium homeostasis that led to arrhythmias at the single-cell level. Mechanistically, we show that the platelet-derived growth factor (PDGF) signalling pathway is activated in mutant iPSC-CMs compared to isogenic control iPSC-CMs. Conversely, pharmacological and molecular inhibition of the PDGF signalling pathway ameliorated the arrhythmic phenotypes of mutant iPSC-CMs in vitro. Taken together, our findings suggest that the activation of the PDGF pathway contributes to the pathogenesis of LMNA-related DCM and point to PDGF receptor-β (PDGFRB) as a potential therapeutic target.
Figures
Comment in
-
PDGF pathway in LMNA-related dilated cardiomyopathy.Nat Rev Cardiol. 2019 Oct;16(10):579. doi: 10.1038/s41569-019-0246-6. Nat Rev Cardiol. 2019. PMID: 31371813 No abstract available.
References
-
- Carmosino M et al. Role of nuclear Lamin A/C in cardiomyocyte functions. Biol. Cell 106, 346–358 (2014). - PubMed
-
- Fatkin D et al. Missense Mutations in the Rod Domain of the Lamin A/C Gene as Causes of Dilated Cardiomyopathy and Conduction-System Disease. New England Journal of Medicine 341, 1715–1724 (1999). - PubMed
-
- Krohne G & Benavente R The nuclear lamins. Experimental Cell Research 162, 1–10 (1986). - PubMed
-
- Hershberger RE & Morales A LMNA-Related Dilated Cardiomyopathy in GeneReviews(®) (eds. Pagon RA et al. ) (University of Washington, Seattle, 1993).
-
- Hershberger RE, Hedges DJ & Morales A Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 10, 531–547 (2013). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- RM1 HG007735/HG/NHGRI NIH HHS/United States
- R01 HL113006/HL/NHLBI NIH HHS/United States
- R00 HL133473/HL/NHLBI NIH HHS/United States
- R01 HL130020/HL/NHLBI NIH HHS/United States
- R01 HL141851/HL/NHLBI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- K99 HL133473/HL/NHLBI NIH HHS/United States
- 17IRG33410532/AHA/American Heart Association-American Stroke Association/United States
- R24 HL117756/HL/NHLBI NIH HHS/United States
- P50 HG007735/HG/NHGRI NIH HHS/United States
- R00 HL104002/HL/NHLBI NIH HHS/United States
- T32 HL094274/HL/NHLBI NIH HHS/United States
- F32 HL139045/HL/NHLBI NIH HHS/United States
- R01 HL132875/HL/NHLBI NIH HHS/United States
- R01 HL139679/HL/NHLBI NIH HHS/United States
- R01 HL128170/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
