

Motor Neuron Susceptibility in ALS/FTD
- PMID: 31316328
- PMCID: PMC6610326
- DOI: 10.3389/fnins.2019.00532
Motor Neuron Susceptibility in ALS/FTD
Abstract
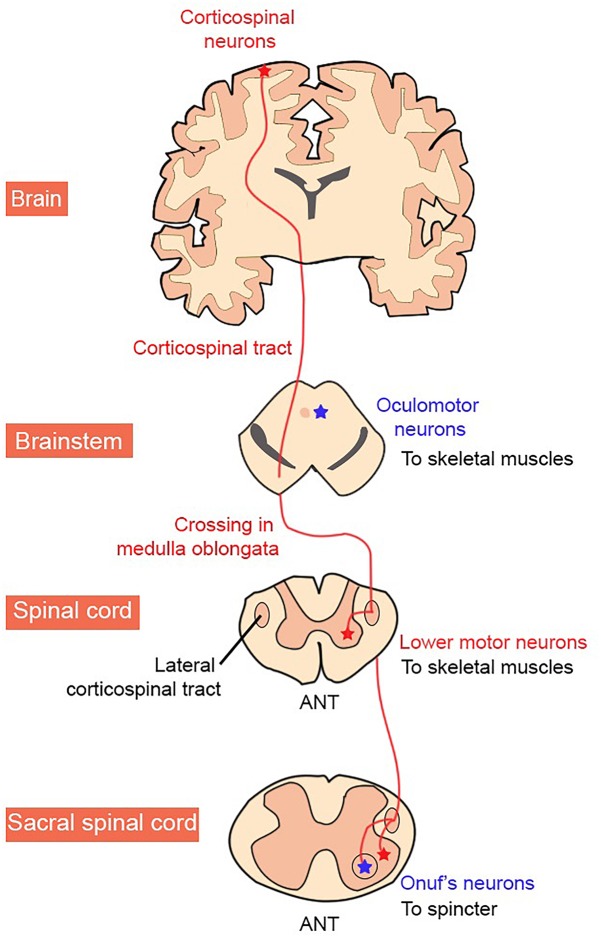
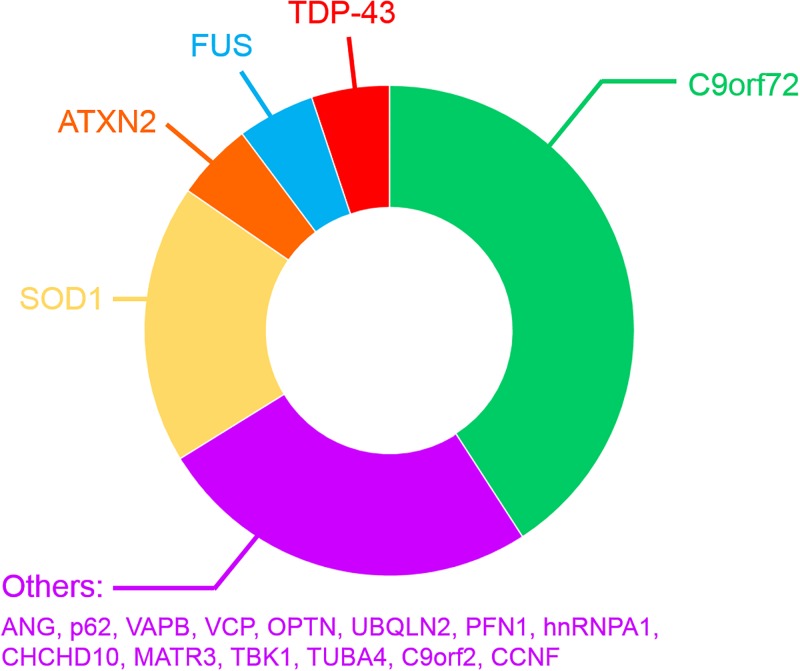
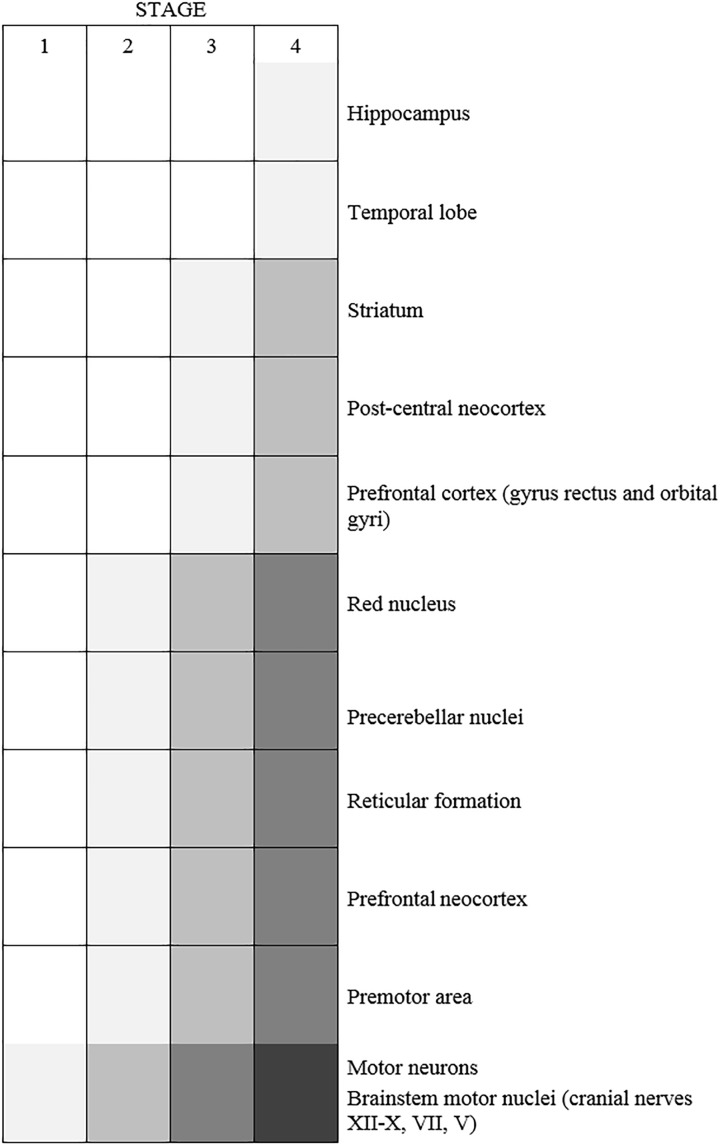
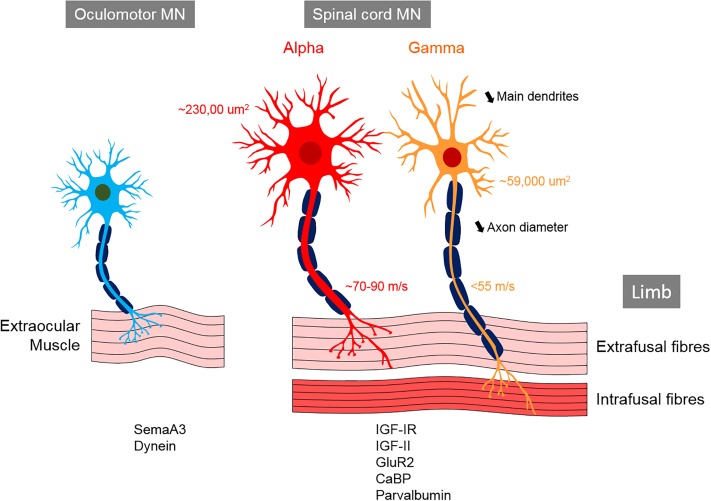
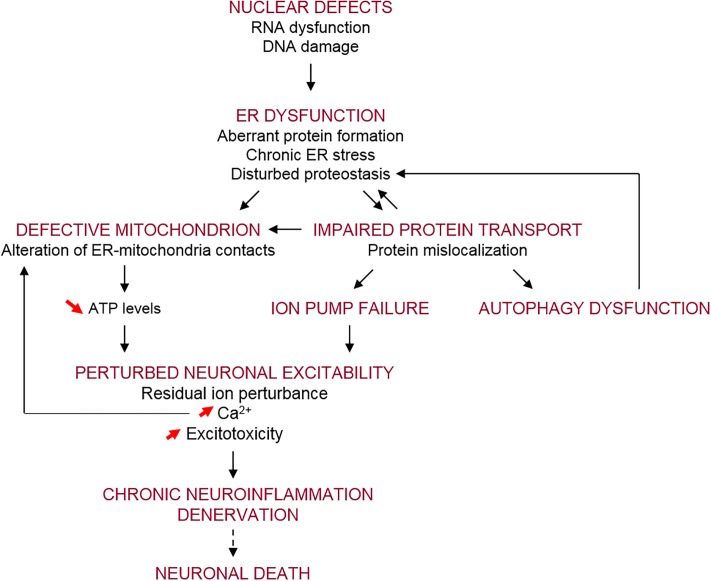
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the death of both upper and lower motor neurons (MNs) in the brain, brainstem and spinal cord. The neurodegenerative mechanisms leading to MN loss in ALS are not fully understood. Importantly, the reasons why MNs are specifically targeted in this disorder are unclear, when the proteins associated genetically or pathologically with ALS are expressed ubiquitously. Furthermore, MNs themselves are not affected equally; specific MNs subpopulations are more susceptible than others in both animal models and human patients. Corticospinal MNs and lower somatic MNs, which innervate voluntary muscles, degenerate more readily than specific subgroups of lower MNs, which remain resistant to degeneration, reflecting the clinical manifestations of ALS. In this review, we discuss the possible factors intrinsic to MNs that render them uniquely susceptible to neurodegeneration in ALS. We also speculate why some MN subpopulations are more vulnerable than others, focusing on both their molecular and physiological properties. Finally, we review the anatomical network and neuronal microenvironment as determinants of MN subtype vulnerability and hence the progression of ALS.
Keywords: amyotrophic lateral sclerosis; fast and slow motor units; frontotemporal dementia; neurodegeneration; selective vulnerability.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous