

Molecular Mechanisms and Therapeutics for the GAA·TTC Expansion Disease Friedreich Ataxia
- PMID: 31317428
- PMCID: PMC6985418
- DOI: 10.1007/s13311-019-00764-x
Molecular Mechanisms and Therapeutics for the GAA·TTC Expansion Disease Friedreich Ataxia
Abstract
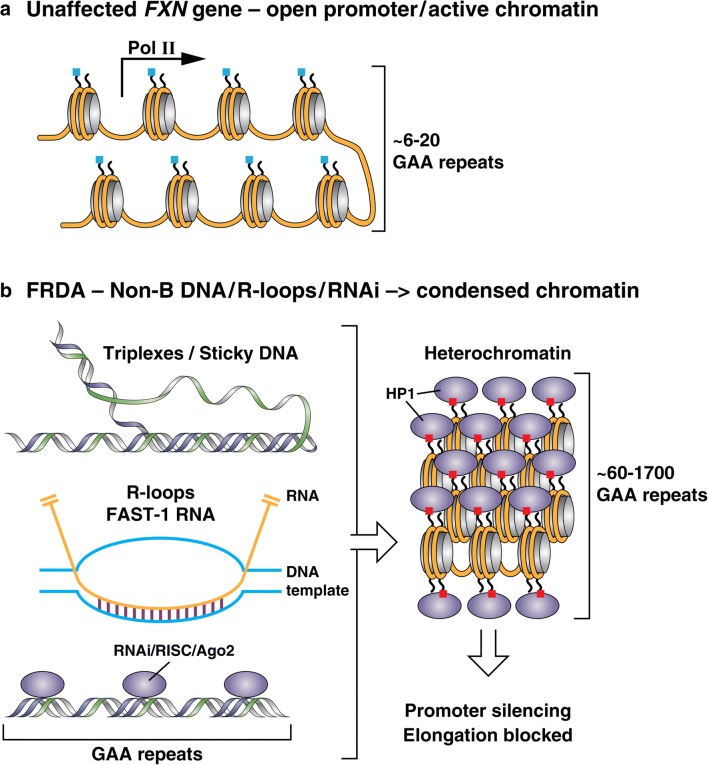
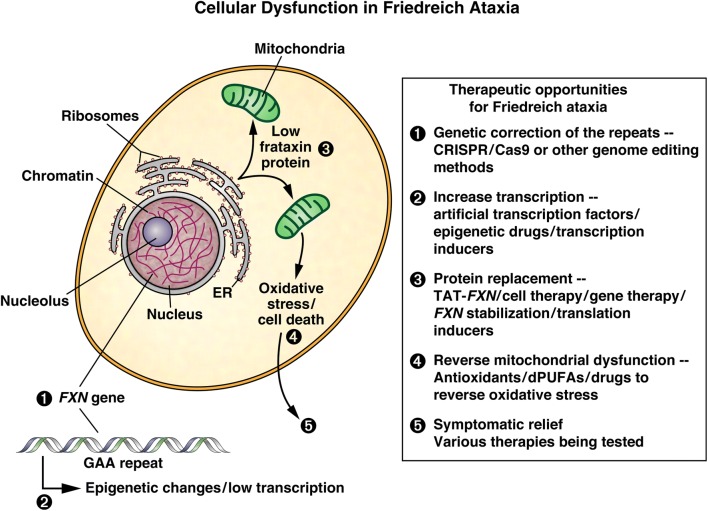
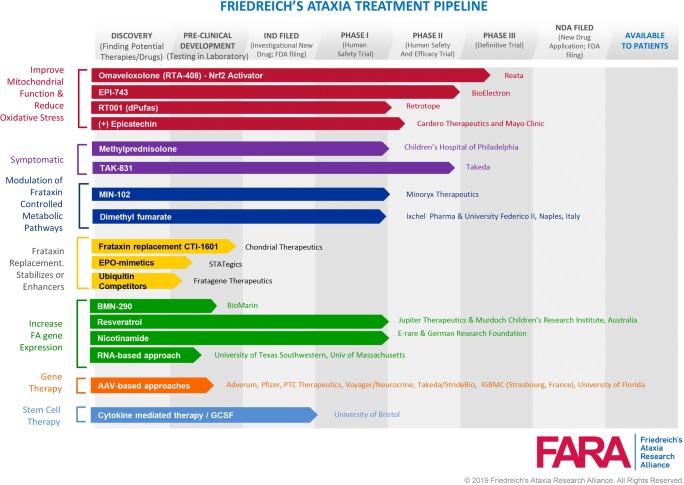
Friedreich ataxia (FRDA), the most common inherited ataxia, is caused by transcriptional silencing of the nuclear FXN gene, encoding the essential mitochondrial protein frataxin. Currently, there is no approved therapy for this fatal disorder. Gene silencing in FRDA is due to hyperexpansion of the triplet repeat sequence GAA·TTC in the first intron of the FXN gene, which results in chromatin histone modifications consistent with heterochromatin formation. Frataxin is involved in mitochondrial iron homeostasis and the assembly and transfer of iron-sulfur clusters to various mitochondrial enzymes and components of the electron transport chain. Frataxin insufficiency leads to progressive spinocerebellar neurodegeneration, causing symptoms of gait and limb ataxia, slurred speech, muscle weakness, sensory loss, and cardiomyopathy in many patients, resulting in death in early adulthood. Numerous approaches are being taken to find a treatment for FRDA, including excision or correction of the repeats by genome engineering methods, gene activation with small molecules or artificial transcription factors, delivery of frataxin to affected cells by protein replacement therapy, gene therapy, or small molecules to increase frataxin protein levels, and therapies aimed at countering the cellular consequences of reduced frataxin. This review will summarize the mechanisms involved in repeat-mediated gene silencing and recent efforts aimed at development of therapeutics.
Keywords: Friedreich ataxia; epigenetics; mitochondrial disease; therapeutics; transcription; trinucleotide repeat expansion.
Conflict of interest statement
The author serves as a consultant to BioMarin Pharmaceutical and is an inventor on patents licensed by The Scripps Research Institute to BioMarin Pharmaceutical.
Figures
References
-
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annual Review of Neuroscience. 2007;30:575–621. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
