

Phenotypic Manifestations of Arrhythmogenic Cardiomyopathy in Children and Adolescents
- PMID: 31319917
- PMCID: PMC7261020
- DOI: 10.1016/j.jacc.2019.05.022
Phenotypic Manifestations of Arrhythmogenic Cardiomyopathy in Children and Adolescents
Abstract
Background: Arrhythmogenic cardiomyopathy (ACM) is a variably penetrant disease increasingly identified in young patients.
Objectives: This study sought to describe the diverse phenotype, genotype, and outcomes in pediatric and adolescent patients.
Methods: Records from 1999 to 2016 were reviewed for individuals age <21 years with a consistent personal or family history. Patients were categorized by right ventricular (RV), left dominant (LD), or biventricular subtypes using 2010 Task Force Criteria or proposed features of LD disease, encompassing electrocardiographic, structural, histological, and arrhythmic characteristics. Genetic variants classified as pathogenic and/or likely pathogenic by 2015 American College of Medical Genetics and Genomics criteria in recognized disease-associated genes were included.
Results: Manifest disease was evident in 32 patients (age 15.1 ± 3.8 years), of whom 22 were probands, including 16 RV, 7 LD, and 9 biventricular ACM. Nondiagnostic features were seen in 5 of 15 family members. RV disease was associated with cardiac arrest and ventricular tachycardia (p = 0.02) and prevalence of PKP2 variants (p < 0.01), whereas biventricular disease was associated with a younger age of onset (p = 0.02). LD ACM was associated with variants in DSP and LMNA, and biventricular ACM with more a diverse etiology in desmosomal genes. Cardiac arrest was observed in 5 probands (age 15.3 ± 1.9 years) and ventricular tachycardia in 10 (age 16.6 ± 2.7 years), 6 probands, and 4 family members. Features suggestive of myocardial inflammation were seen in 6 patients, with ventricular tachycardia and/or cardiac arrest in 3 patients. Cardiac transplantation was performed in 10 patients. There were no deaths. In RV and biventricular disease, electrocardiographic preceded imaging features, whereas the reverse was seen in LD disease.
Conclusions: ACM in the young has highly varied phenotypic expression incorporating life-threatening arrhythmia, heart failure, and myocardial inflammation. Increased awareness of early onset, aggressive disease has important implications for patient management and familial screening.
Keywords: arrhythmogenic right ventricular cardiomyopathy; desmosomes; diagnostic criteria; genetics; pediatrics; phenotype.
Copyright © 2019 American College of Cardiology Foundation. Published by Elsevier Inc. All rights reserved.
Figures


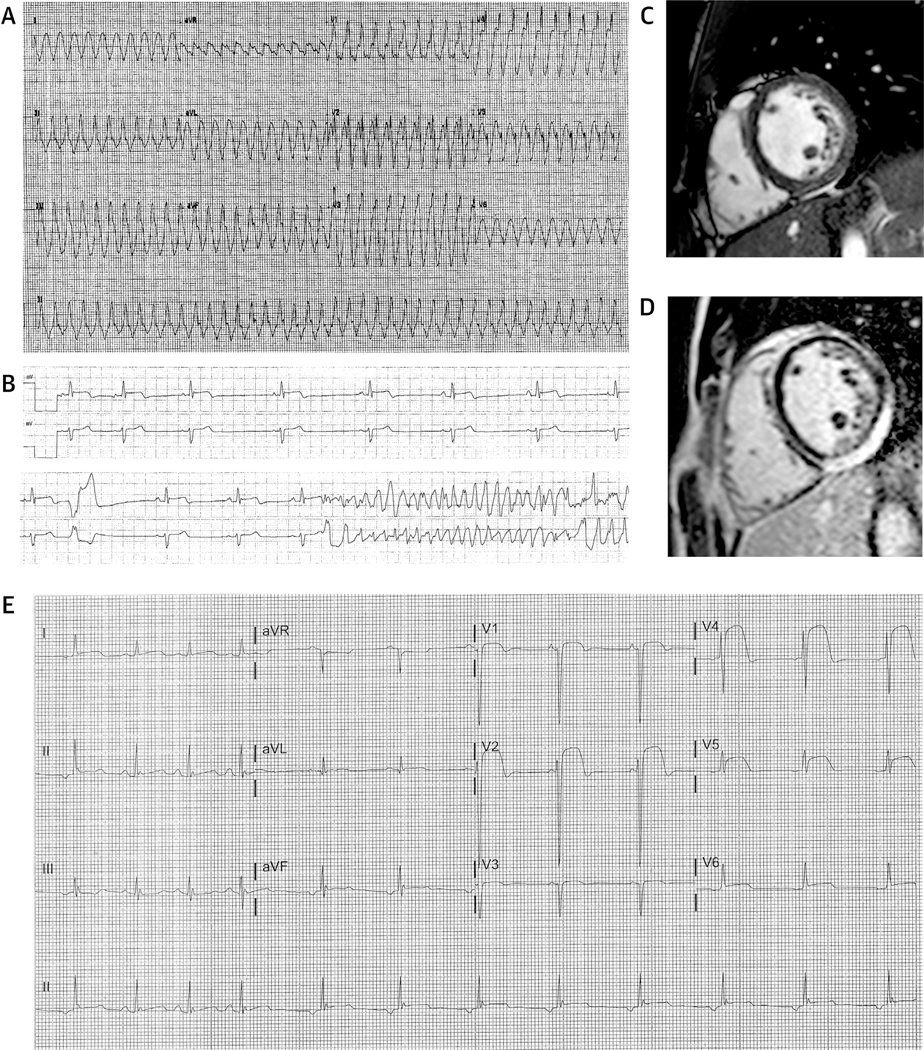

Comment in
-
Pediatric-Onset Arrhythmogenic Cardiomyopathy: Look Right, Look Left, Look Both Ways.J Am Coll Cardiol. 2019 Jul 23;74(3):359-361. doi: 10.1016/j.jacc.2019.05.023. J Am Coll Cardiol. 2019. PMID: 31319918 No abstract available.
References
-
- Sen-Chowdhry S, McKenna WJ. Reconciling the protean manifestations of arrhythmogenic cardiomyopathy. Circ Arrhythm Electrophysiol 2010; 3:566–70. - PubMed
-
- Marcus FI, Fontaine GH, Guiraudon G, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation 1982;65:384–98. - PubMed
-
- Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007;115:1710–20. - PubMed
-
- van Tintelen JP, Van Gelder IC, Asimaki A, et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm 2009;6:1574–83. - PubMed
-
- Quarta G, Syrris P, Ashworth M, et al. Mutations in the lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy.EurHeart J 2012;33:1128–36. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
