

Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets
- PMID: 31320640
- PMCID: PMC6639323
- DOI: 10.1038/s41467-019-11107-x
Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets
Abstract
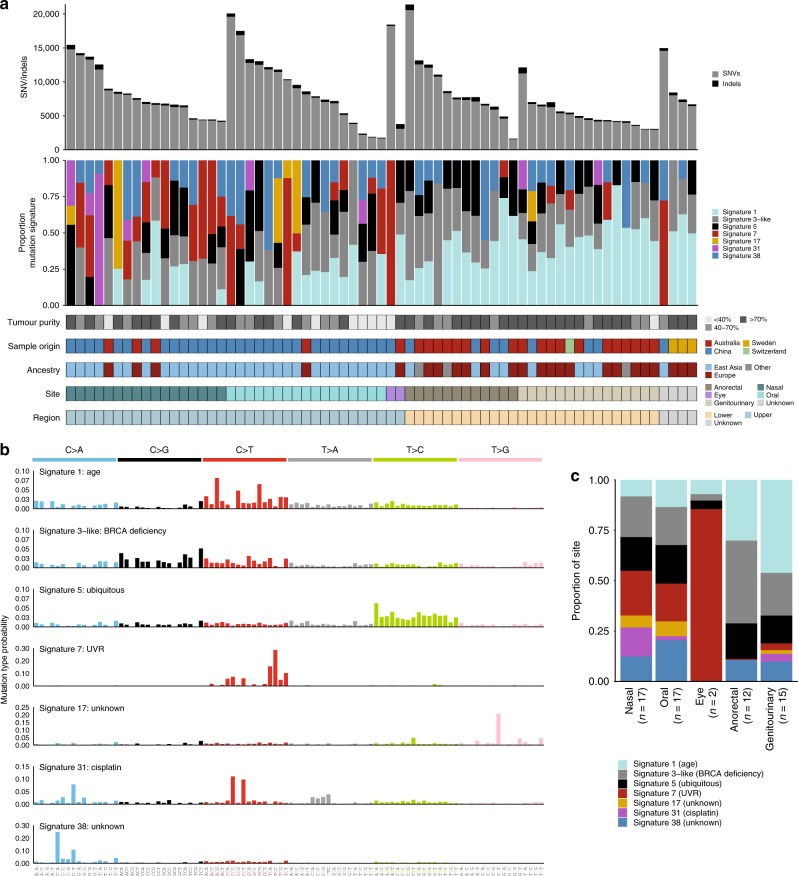
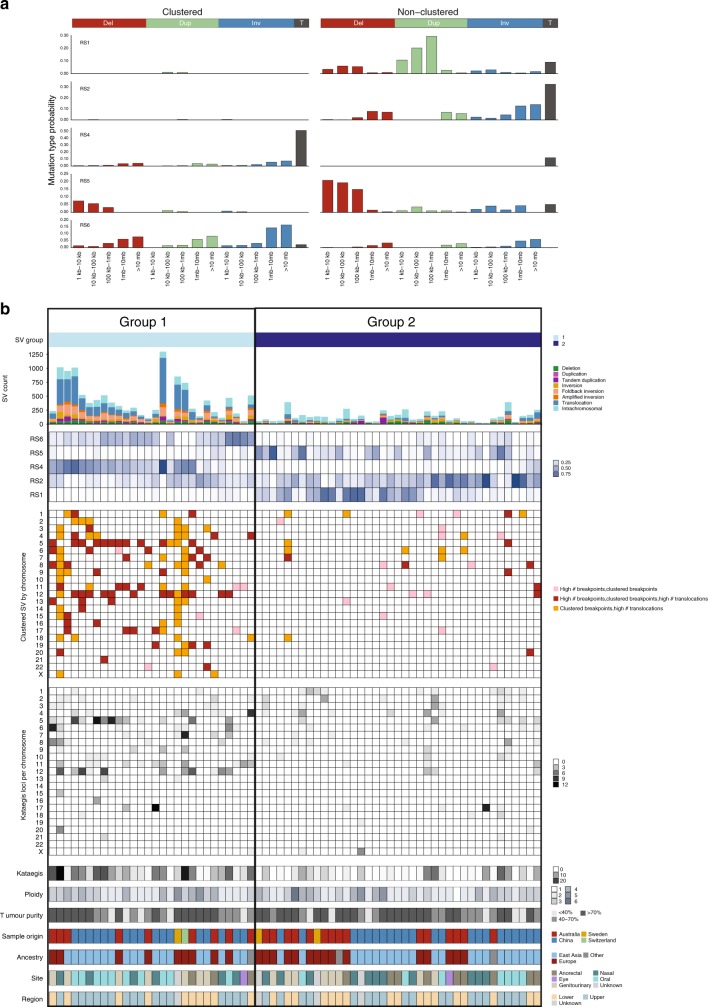
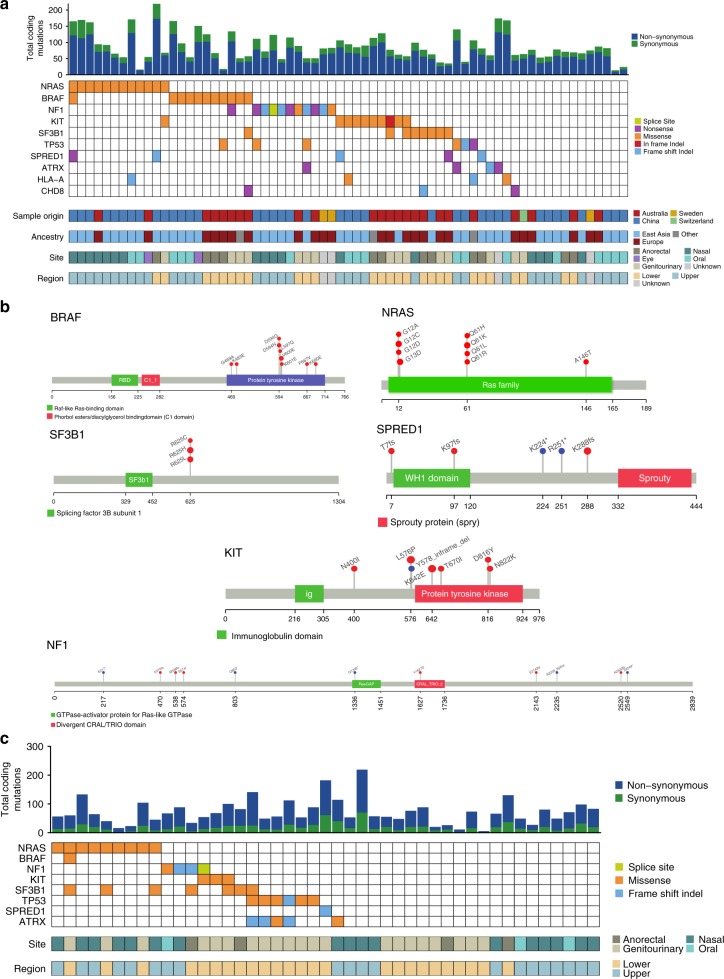
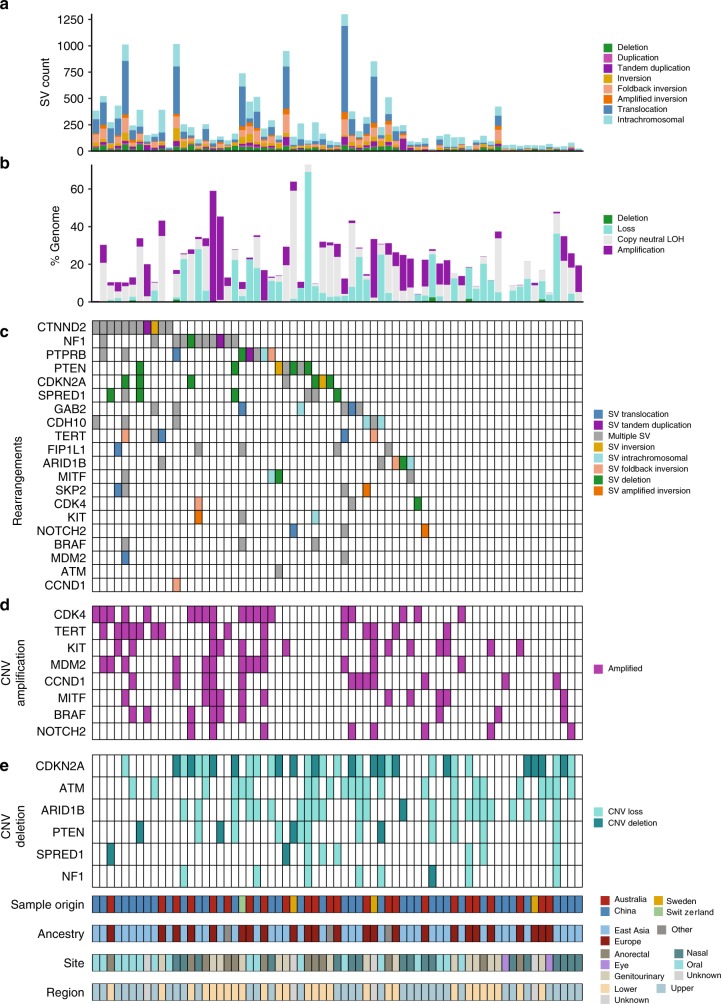
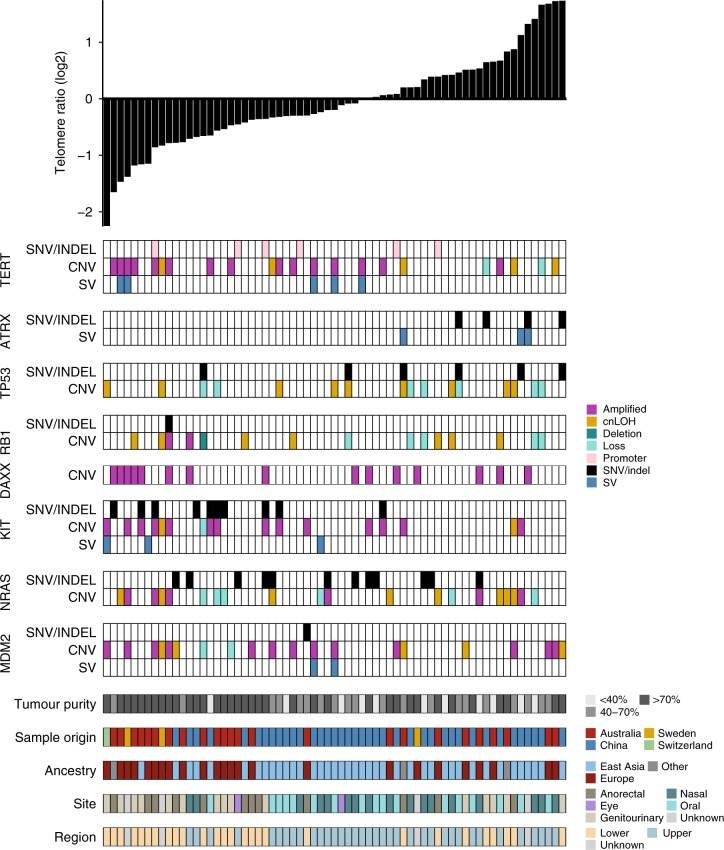
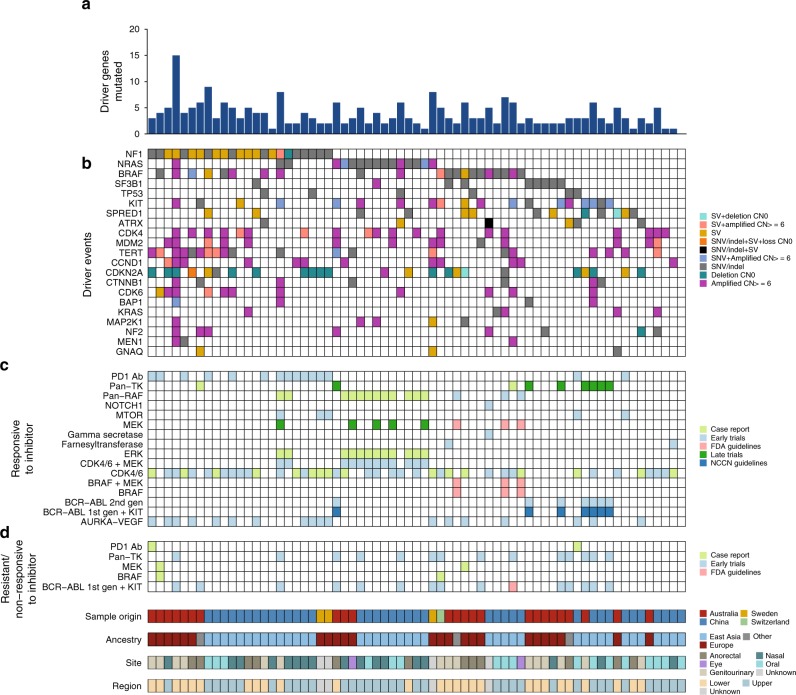
Knowledge of key drivers and therapeutic targets in mucosal melanoma is limited due to the paucity of comprehensive mutation data on this rare tumor type. To better understand the genomic landscape of mucosal melanoma, here we describe whole genome sequencing analysis of 67 tumors and validation of driver gene mutations by exome sequencing of 45 tumors. Tumors have a low point mutation burden and high numbers of structural variants, including recurrent structural rearrangements targeting TERT, CDK4 and MDM2. Significantly mutated genes are NRAS, BRAF, NF1, KIT, SF3B1, TP53, SPRED1, ATRX, HLA-A and CHD8. SF3B1 mutations occur more commonly in female genital and anorectal melanomas and CTNNB1 mutations implicate a role for WNT signaling defects in the genesis of some mucosal melanomas. TERT aberrations and ATRX mutations are associated with alterations in telomere length. Mutation profiles of the majority of mucosal melanomas suggest potential susceptibility to CDK4/6 and/or MEK inhibitors.
Conflict of interest statement
J.V.P. and N.W. are founders and shareholders of genomiQa Pty Ltd, and members of its Board. B.C.B. is a Consultant to Lilly Inc. D.J.A. is a paid consultant for Microbiotica and receives research funding support from OpenTargets and BMS. None of these relationships involve the work described in this manuscript. R.D. has intermittent, project focused consulting and/or advisory relationships with Novartis, Merck Sharp & Dhome (MSD), Bristol-Myers Squibb (BMS), Roche, Amgen, Takeda, Pierre Fabre, Sun Pharma, Sanofi outside the submitted work. J.G. is consultant of MSD, Novartis, Pfizer and Bayer. J.F.T. is a Scientific Advisory Board member for GlaxoSmithKline, BMS, MSD Australia, and Provectus Inc. R.A.S. is a Scientific Advisor, Board Member for Merck Sharp & Dhome (MSD) and Novartis. None of these relationships involve the work described in this manuscript. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
