

Mechanistic inferences on metabolic dysfunction in posttraumatic stress disorder from an integrated model and multiomic analysis: role of glucocorticoid receptor sensitivity
- PMID: 31322414
- PMCID: PMC6879860
- DOI: 10.1152/ajpendo.00065.2019
Mechanistic inferences on metabolic dysfunction in posttraumatic stress disorder from an integrated model and multiomic analysis: role of glucocorticoid receptor sensitivity
Abstract
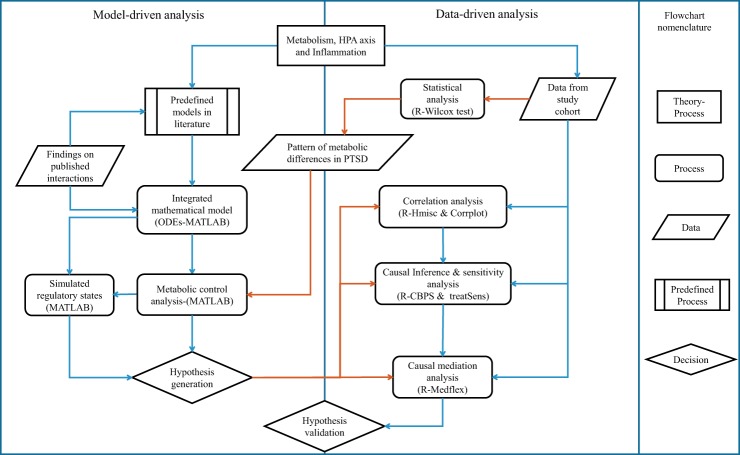
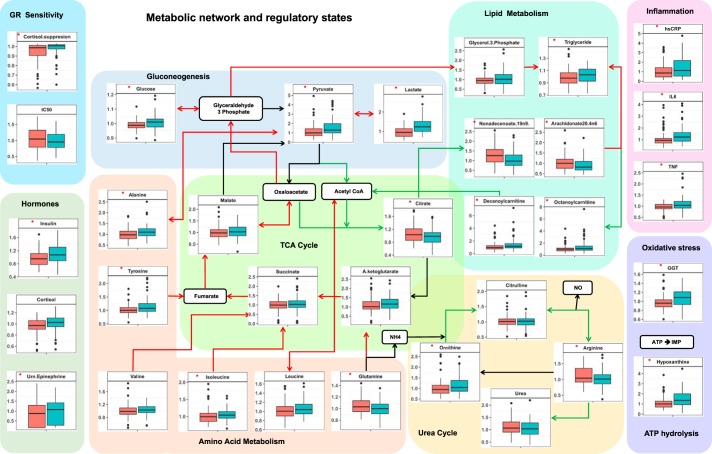
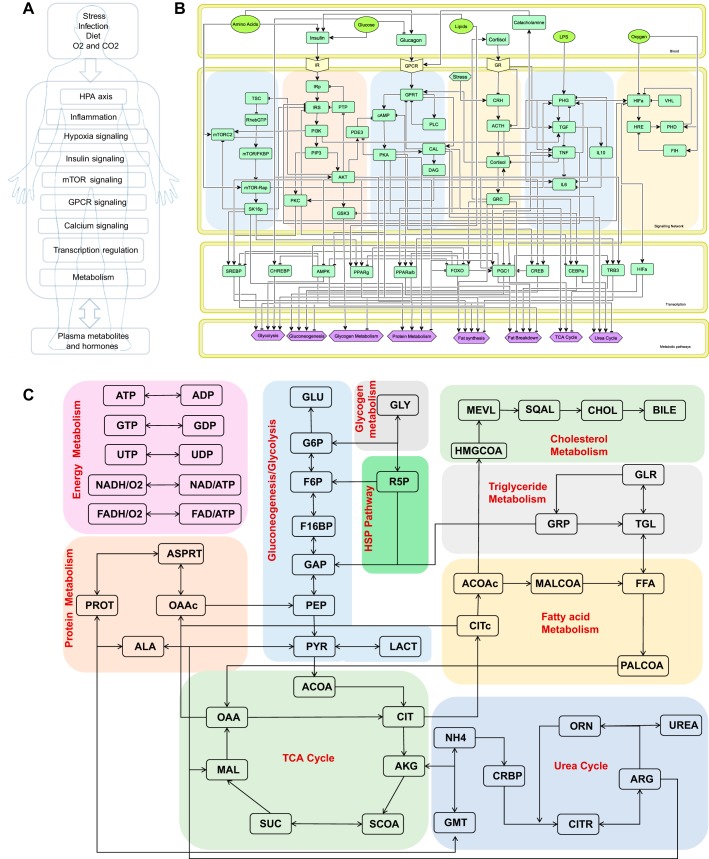
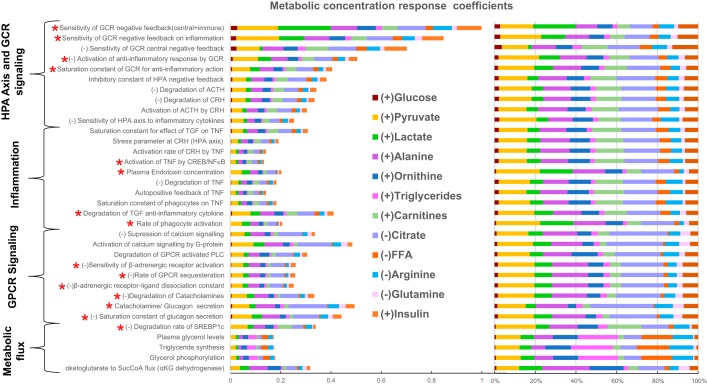
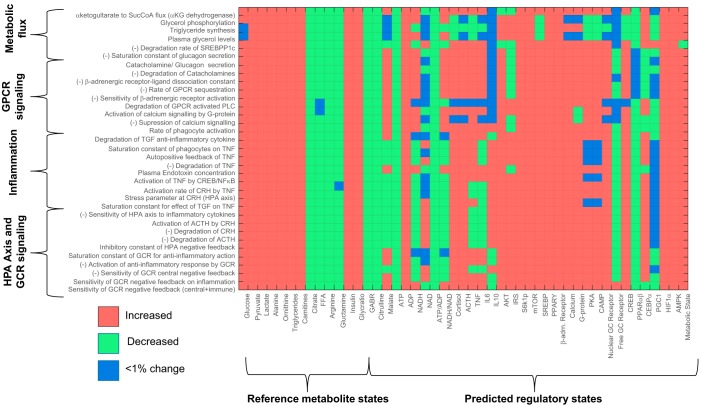
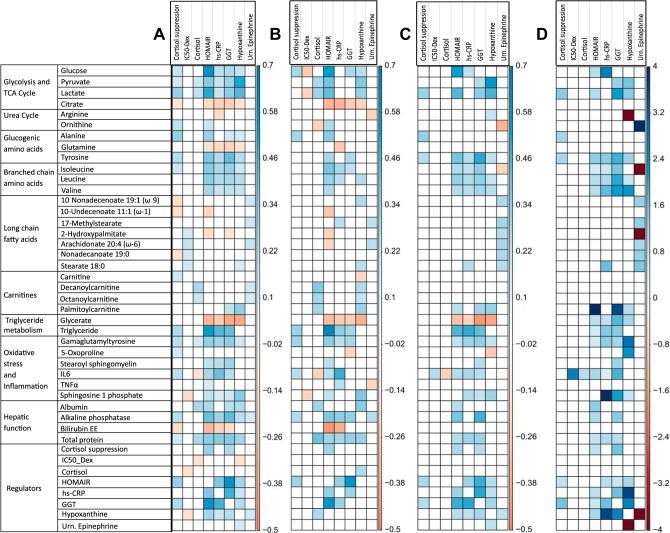
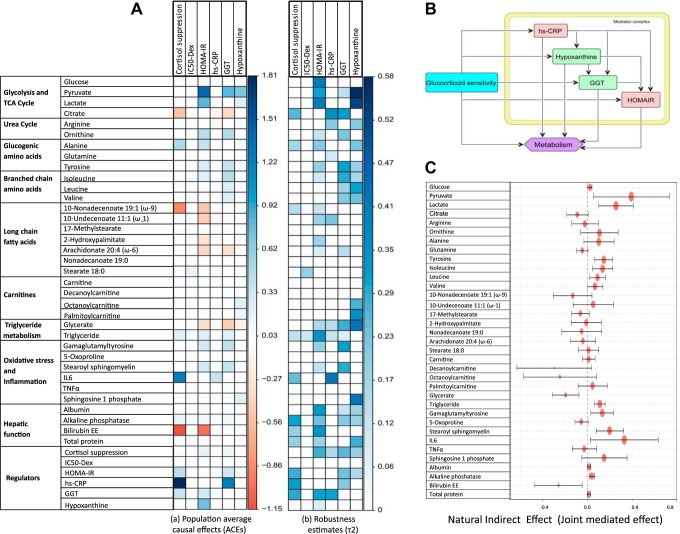
Posttraumatic stress disorder (PTSD) is associated with neuroendocrine alterations and metabolic abnormalities; however, how metabolism is affected by neuroendocrine disturbances is unclear. The data from combat-exposed veterans with PTSD show increased glycolysis to lactate flux, reduced TCA cycle flux, impaired amino acid and lipid metabolism, insulin resistance, inflammation, and hypersensitive hypothalamic-pituitary-adrenal (HPA) axis. To analyze whether the co-occurrence of multiple metabolic abnormalities is independent or arises from an underlying regulatory defect, we employed a systems biological approach using an integrated mathematical model and multiomic analysis. The models for hepatic metabolism, HPA axis, inflammation, and regulatory signaling were integrated to perform metabolic control analysis (MCA) with respect to the observations from our clinical data. We combined the metabolomics, neuroendocrine, clinical laboratory, and cytokine data from combat-exposed veterans with and without PTSD to characterize the differences in regulatory effects. MCA revealed mechanistic association of the HPA axis and inflammation with metabolic dysfunction consistent with PTSD. This was supported by the data using correlational and causal analysis that revealed significant associations between cortisol suppression, high-sensitivity C-reactive protein, homeostatic model assessment of insulin resistance, γ-glutamyltransferase, hypoxanthine, and several metabolites. Causal mediation analysis indicates that the effects of enhanced glucocorticoid receptor sensitivity (GRS) on glycolytic pathway, gluconeogenic and branched-chain amino acids, triglycerides, and hepatic function are jointly mediated by inflammation, insulin resistance, oxidative stress, and energy deficit. Our analysis suggests that the interventions to normalize GRS and inflammation may help to manage features of metabolic dysfunction in PTSD.
Keywords: HPA axis; PTSD; glucocorticoid signaling; mathematical modeling; neuroendocrine.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
References
-
- American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders (5th ed.). Washington, DC: American Psychiatric Association, 2013. doi:10.1176/appi.books.9780890425596. - DOI
-
- Apostolopoulou M, Gordillo R, Koliaki C, Gancheva S, Jelenik T, De Filippo E, Herder C, Markgraf D, Jankowiak F, Esposito I, Schlensak M, Scherer PE, Roden M. Specific hepatic sphingolipids relate to insulin resistance, oxidative stress, and inflammation in nonalcoholic steatohepatitis. Diabetes Care 41: 1235–1243, 2018. doi:10.2337/dc17-1318. - DOI - PubMed
-
- Bersani FS, Morley C, Lindqvist D, Epel ES, Picard M, Yehuda R, Flory J, Bierer LM, Makotkine I, Abu-Amara D, Coy M, Reus VI, Lin J, Blackburn EH, Marmar C, Wolkowitz OM, Mellon SH. Mitochondrial DNA copy number is reduced in male combat veterans with PTSD. Prog Neuropsychopharmacol Biol Psychiatry 64: 10–17, 2016. doi:10.1016/j.pnpbp.2015.06.012. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials