

Induction of Acquired Resistance towards EGFR Inhibitor Gefitinib in a Patient-Derived Xenograft Model of Non-Small Cell Lung Cancer and Subsequent Molecular Characterization
- PMID: 31323891
- PMCID: PMC6678194
- DOI: 10.3390/cells8070740
Induction of Acquired Resistance towards EGFR Inhibitor Gefitinib in a Patient-Derived Xenograft Model of Non-Small Cell Lung Cancer and Subsequent Molecular Characterization
Abstract
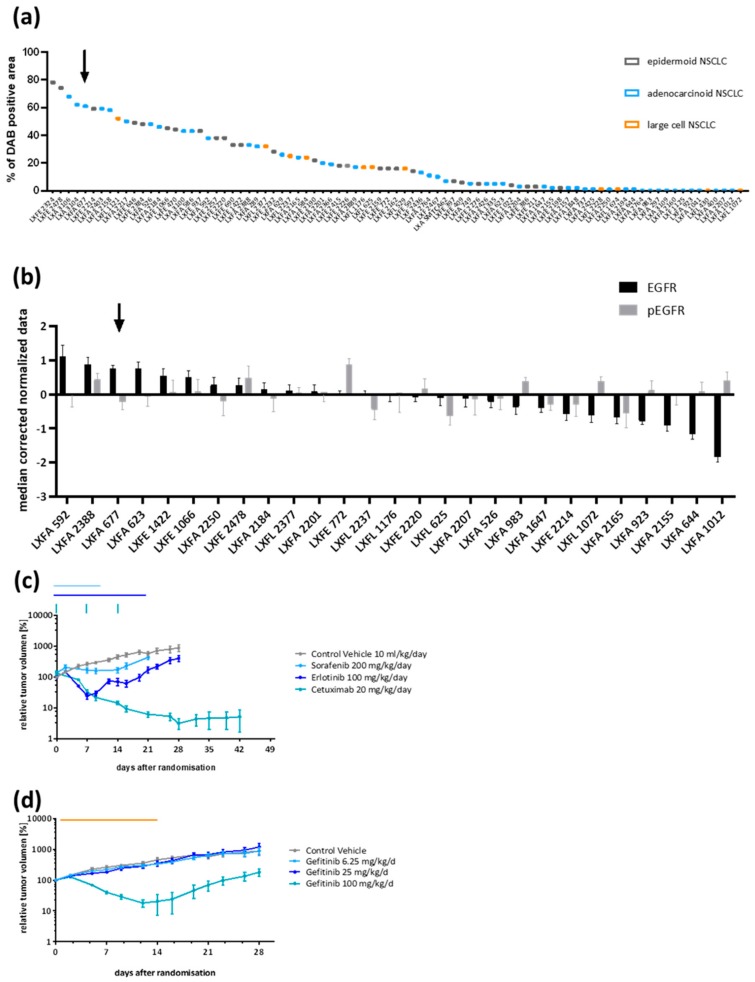
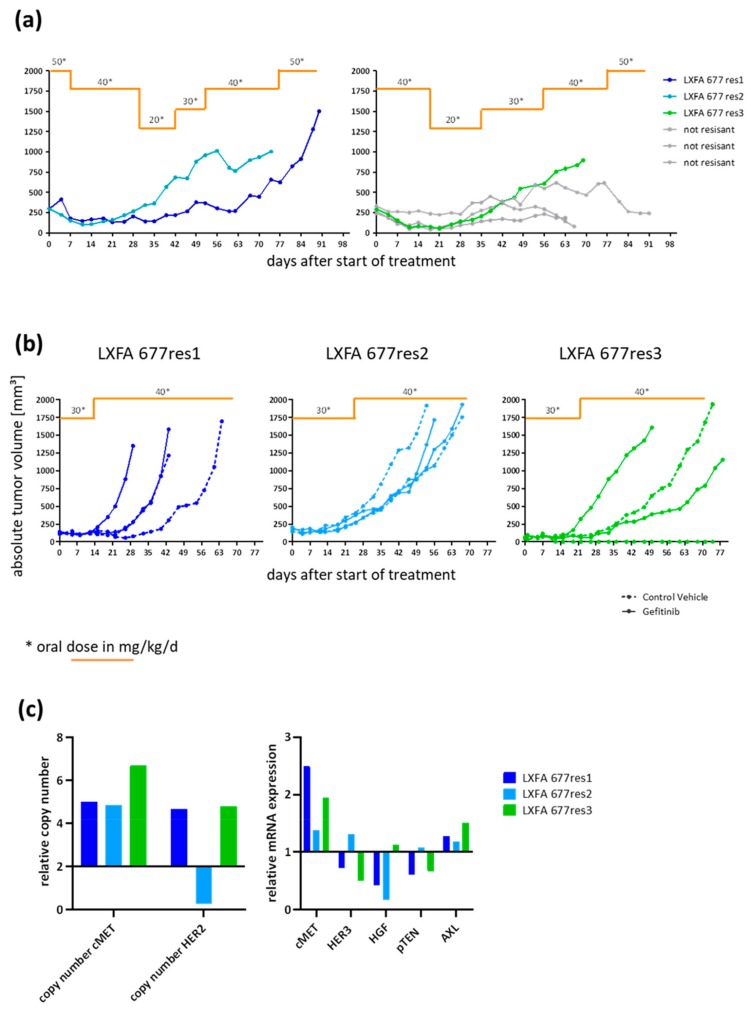
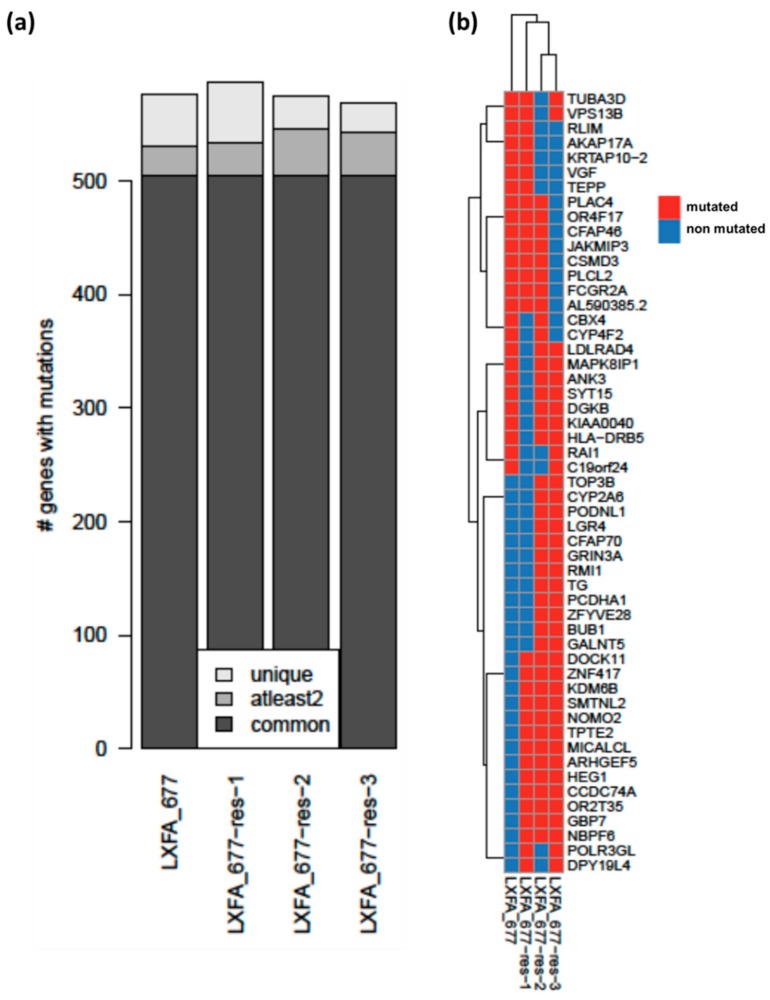
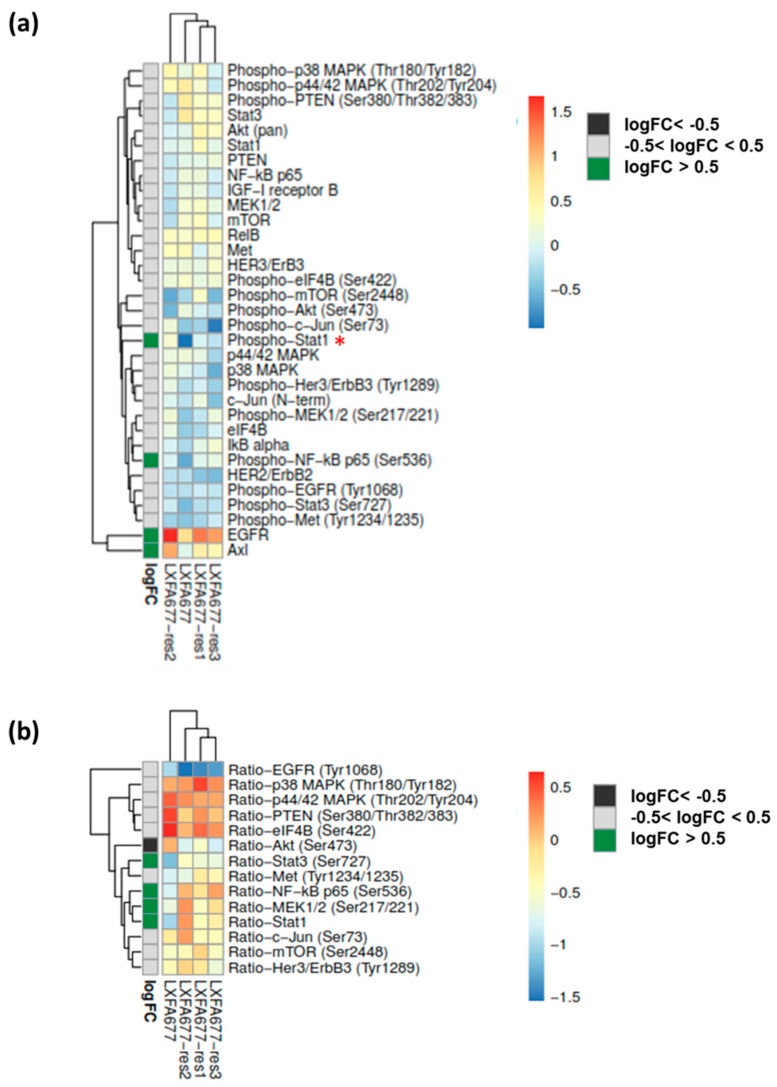
In up to 30% of non-small cell lung cancer (NSCLC) patients, the oncogenic driver of tumor growth is a constitutively activated epidermal growth factor receptor (EGFR). Although these patients gain great benefit from treatment with EGFR tyrosine kinase inhibitors, the development of resistance is inevitable. To model the emergence of drug resistance, an EGFR-driven, patient-derived xenograft (PDX) NSCLC model was treated continuously with Gefitinib in vivo. Over a period of more than three months, three separate clones developed and were subsequently analyzed: Whole exome sequencing and reverse phase protein arrays (RPPAs) were performed to identify the mechanism of resistance. In total, 13 genes were identified, which were mutated in all three resistant lines. Amongst them the mutations in NOMO2, ARHGEF5 and SMTNL2 were predicted as deleterious. The 53 mutated genes specific for at least two of the resistant lines were mainly involved in cell cycle activities or the Fanconi anemia pathway. On a protein level, total EGFR, total Axl, phospho-NFκB, and phospho-Stat1 were upregulated. Stat1, Stat3, MEK1/2, and NFκB displayed enhanced activation in the resistant clones determined by the phosphorylated vs. total protein ratio. In summary, we developed an NSCLC PDX line modelling possible escape mechanism under EGFR treatment. We identified three genes that have not been described before to be involved in an acquired EGFR resistance. Further functional studies are needed to decipher the underlying pathway regulation.
Keywords: EGFR inhibition; NSCLC; PDX; acquired resistance; reverse phase protein array; whole exome sequencing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Wang Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017;1652:3–35. - PubMed
-
- Siu M.K., Abou-Kheir W., Yin J.J., Chang Y.S., Barrett B., Suau F., Casey O., Chen W.Y., Fang L., Hynes P., et al. Loss of EGFR signaling regulated miR-203 promotes prostate cancer bone metastasis and tyrosine kinase inhibitors resistance. Oncotarget. 2014;5:3770–3784. doi: 10.18632/oncotarget.1994. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
