

Genetic mapping of cell type specificity for complex traits
- PMID: 31324783
- PMCID: PMC6642112
- DOI: 10.1038/s41467-019-11181-1
Genetic mapping of cell type specificity for complex traits
Erratum in
-
Author Correction: Genetic mapping of cell type specificity for complex traits.Nat Commun. 2020 Apr 1;11(1):1718. doi: 10.1038/s41467-020-15365-y. Nat Commun. 2020. PMID: 32238815 Free PMC article.
Abstract
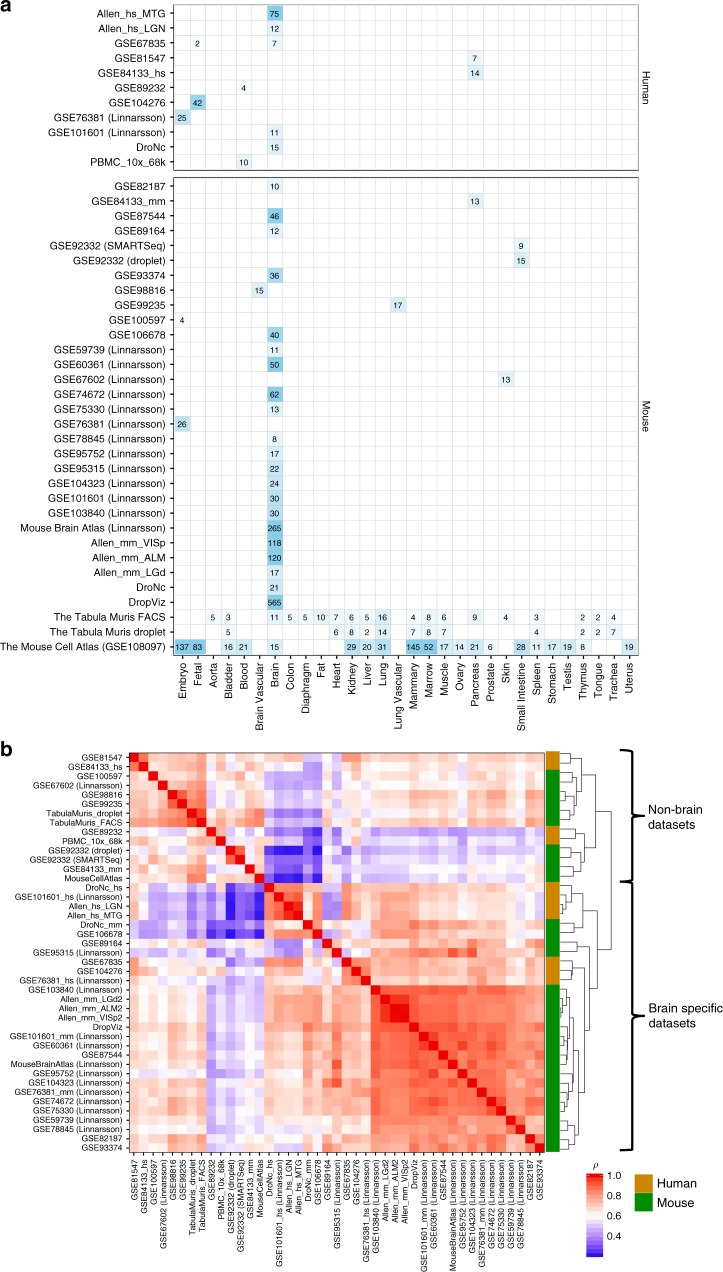
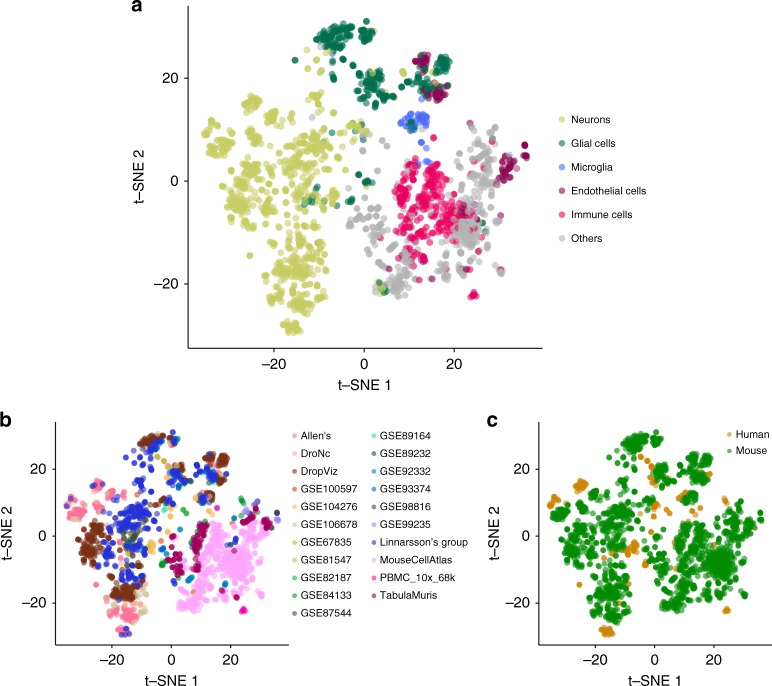
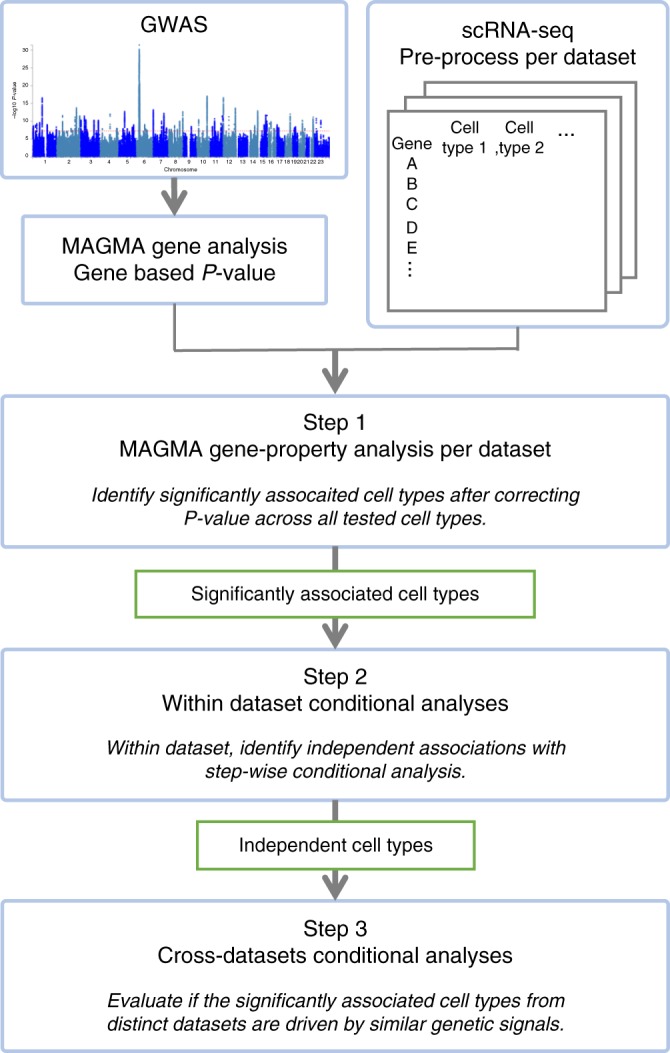
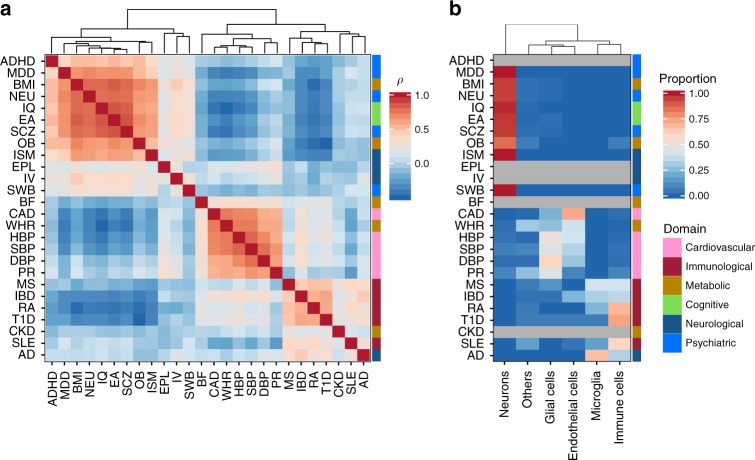
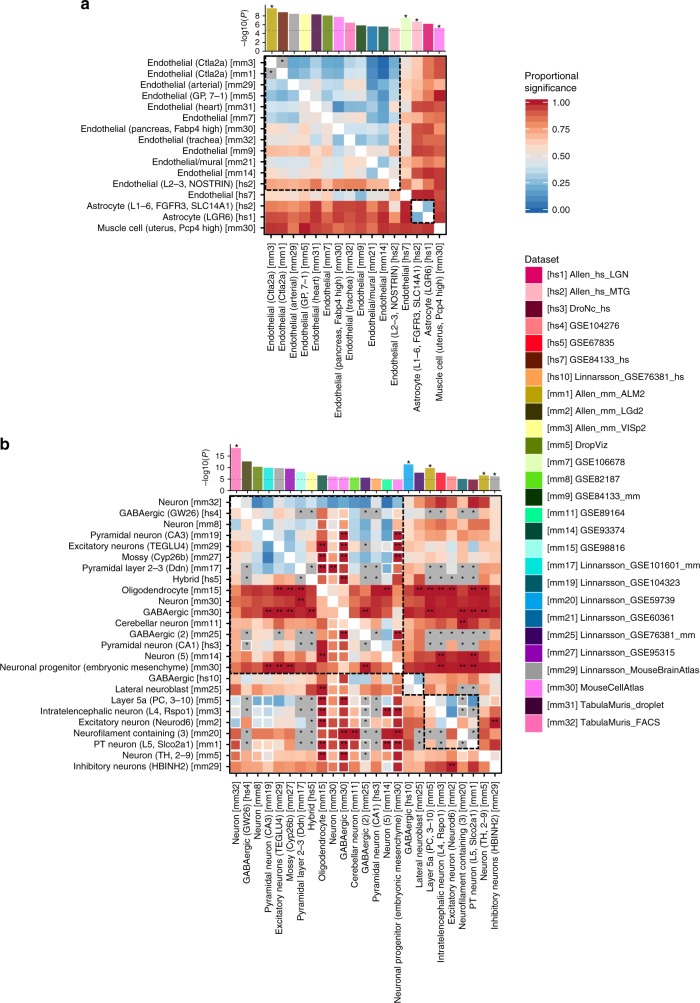
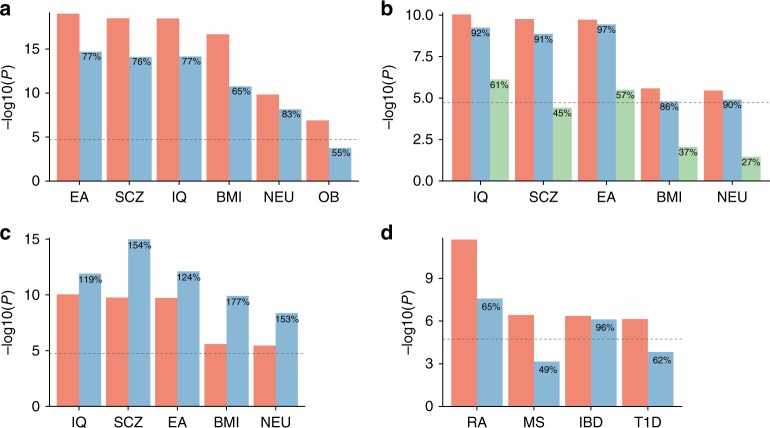
Single-cell RNA sequencing (scRNA-seq) data allows to create cell type specific transcriptome profiles. Such profiles can be aligned with genome-wide association studies (GWASs) to implicate cell type specificity of the traits. Current methods typically rely only on a small subset of available scRNA-seq datasets, and integrating multiple datasets is hampered by complex batch effects. Here we collated 43 publicly available scRNA-seq datasets. We propose a 3-step workflow with conditional analyses within and between datasets, circumventing batch effects, to uncover associations of traits with cell types. Applying this method to 26 traits, we identify independent associations of multiple cell types. These results lead to starting points for follow-up functional studies aimed at gaining a mechanistic understanding of these traits. The proposed framework as well as the curated scRNA-seq datasets are made available via an online platform, FUMA, to facilitate rapid evaluation of cell type specificity by other researchers.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
