

ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes
- PMID: 31329164
- PMCID: PMC6668702
- DOI: 10.1172/JCI125316
ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes
Abstract
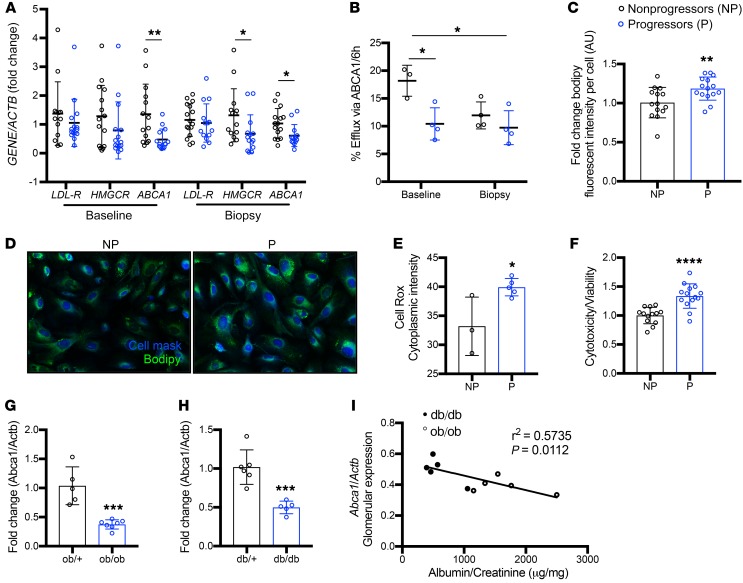
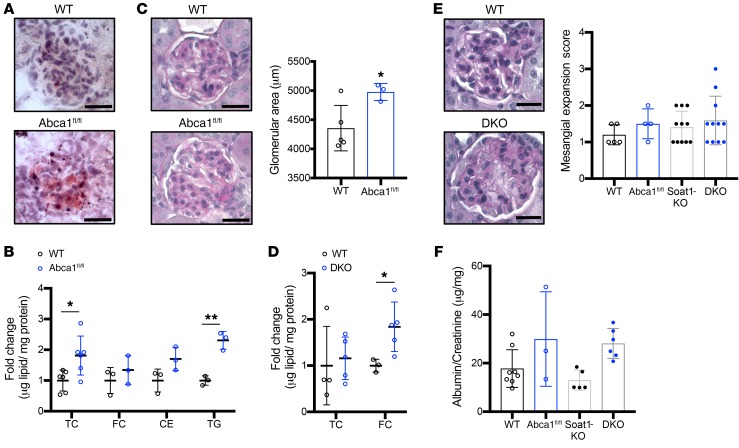
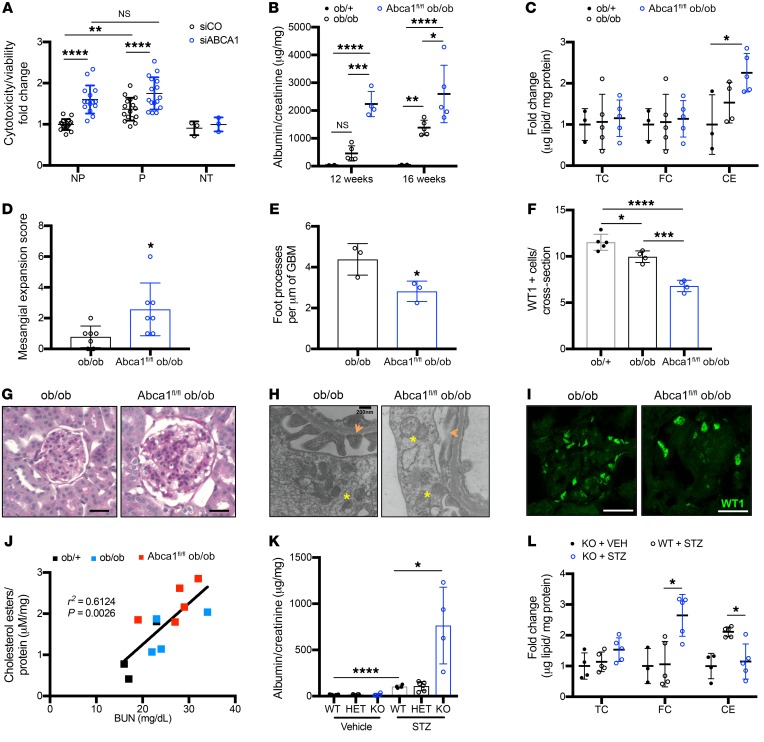
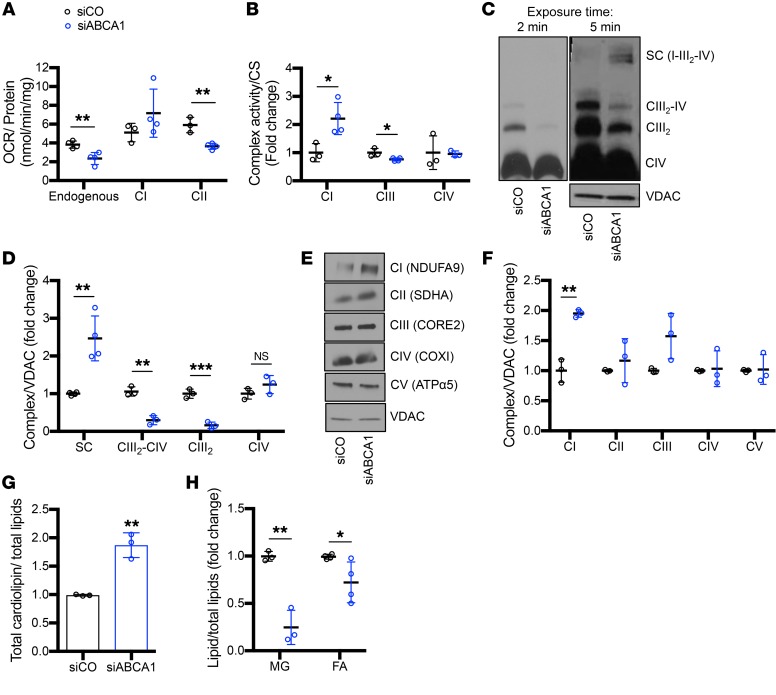
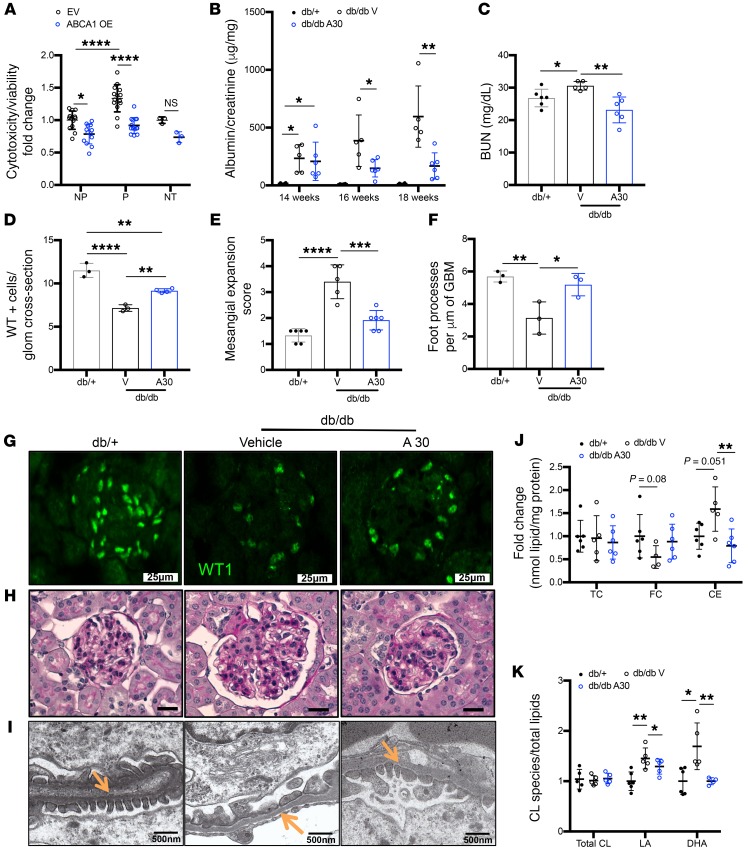
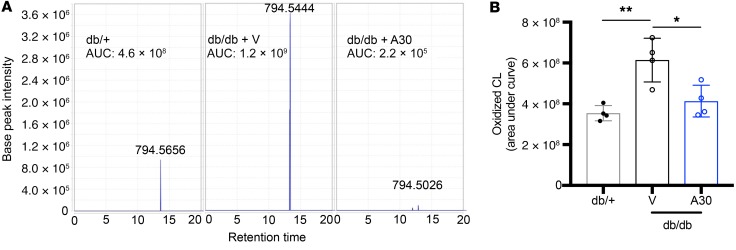
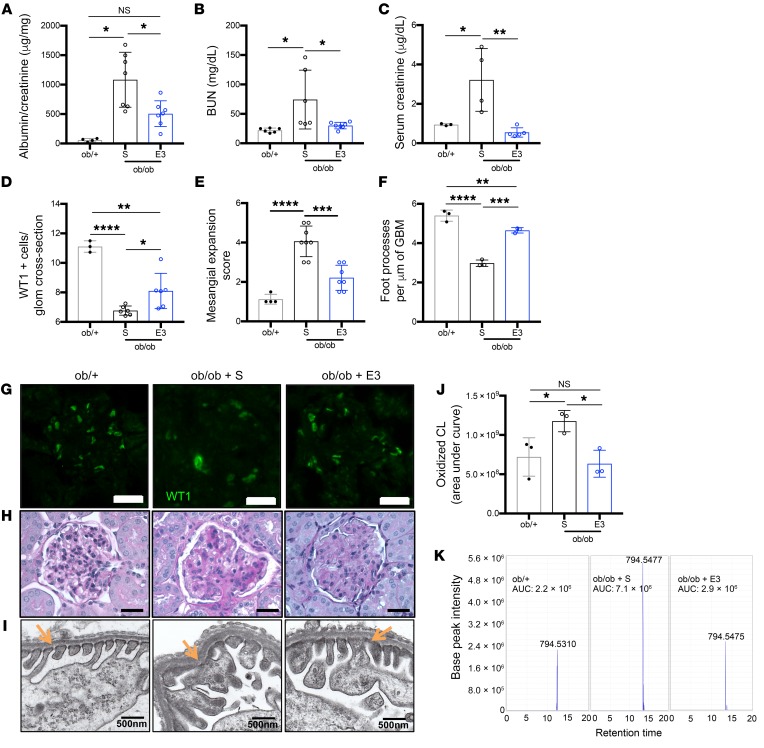
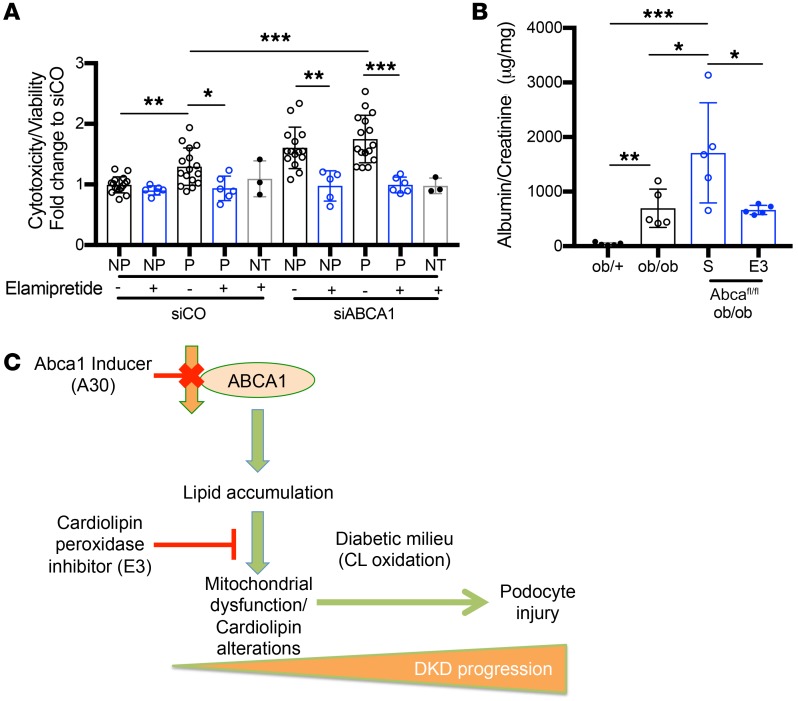
Fibroblasts from patients with Tangier disease carrying ATP-binding cassette A1 (ABCA1) loss-of-function mutations are characterized by cardiolipin accumulation, a mitochondrial-specific phospholipid. Suppression of ABCA1 expression occurs in glomeruli from patients with diabetic kidney disease (DKD) and in human podocytes exposed to DKD sera collected prior to the development of DKD. We demonstrated that siRNA ABCA1 knockdown in podocytes led to reduced oxygen consumption capabilities associated with alterations in the oxidative phosphorylation (OXPHOS) complexes and with cardiolipin accumulation. Podocyte-specific deletion of Abca1 (Abca1fl/fl) rendered mice susceptible to DKD, and pharmacological induction of ABCA1 improved established DKD. This was not mediated by free cholesterol, as genetic deletion of sterol-o-acyltransferase-1 (SOAT1) in Abca1fl/fl mice was sufficient to cause free cholesterol accumulation but did not cause glomerular injury. Instead, cardiolipin mediates ABCA1-dependent susceptibility to podocyte injury, as inhibition of cardiolipin peroxidation with elamipretide improved DKD in vivo and prevented ABCA1-dependent podocyte injury in vitro and in vivo. Collectively, we describe a pathway definitively linking ABCA1 deficiency to cardiolipin-driven mitochondrial dysfunction. We demonstrated that this pathway is relevant to DKD and that ABCA1 inducers or inhibitors of cardiolipin peroxidation may each represent therapeutic strategies for the treatment of established DKD.
Keywords: Cholesterol; Chronic kidney disease; Metabolism; Mitochondria; Nephrology.
Conflict of interest statement
Figures
Comment in
-
Lipids mediate podocyte damage.Nat Rev Nephrol. 2019 Oct;15(10):594. doi: 10.1038/s41581-019-0196-6. Nat Rev Nephrol. 2019. PMID: 31399727 No abstract available.
-
Podocyte lipotoxicity in diabetic kidney disease.Kidney Int. 2019 Oct;96(4):809-812. doi: 10.1016/j.kint.2019.07.013. Kidney Int. 2019. PMID: 31543147 No abstract available.
References
-
- US Renal Data System 2016 Annual Data Report: Epidemiology of Kidney Disease in the United States Am J Kidney Dis. 2017;69(3S1):A4 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
