

AKR1D1 is a novel regulator of metabolic phenotype in human hepatocytes and is dysregulated in non-alcoholic fatty liver disease
- PMID: 31330134
- PMCID: PMC6744372
- DOI: 10.1016/j.metabol.2019.153947
AKR1D1 is a novel regulator of metabolic phenotype in human hepatocytes and is dysregulated in non-alcoholic fatty liver disease
Abstract
Objective: Non-alcoholic fatty liver disease (NAFLD) is the hepatic manifestation of metabolic syndrome. Steroid hormones and bile acids are potent regulators of hepatic carbohydrate and lipid metabolism. Steroid 5β-reductase (AKR1D1) is highly expressed in human liver where it inactivates steroid hormones and catalyzes a fundamental step in bile acid synthesis.
Methods: Human liver biopsies were obtained from 34 obese patients and AKR1D1 mRNA expression levels were measured using qPCR. Genetic manipulation of AKR1D1 was performed in human HepG2 and Huh7 liver cell lines. Metabolic assessments were made using transcriptome analysis, western blotting, mass spectrometry, clinical biochemistry, and enzyme immunoassays.
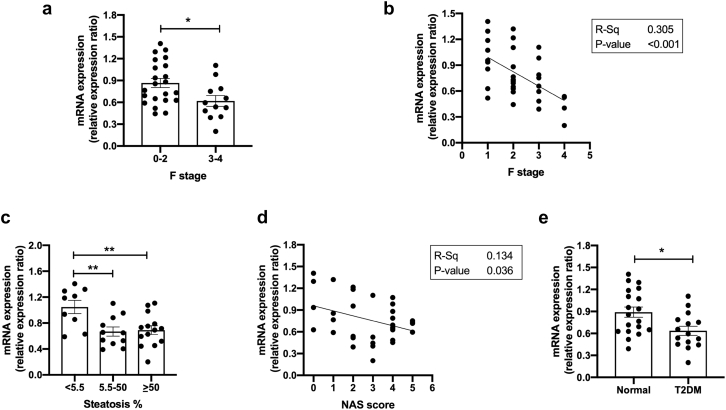
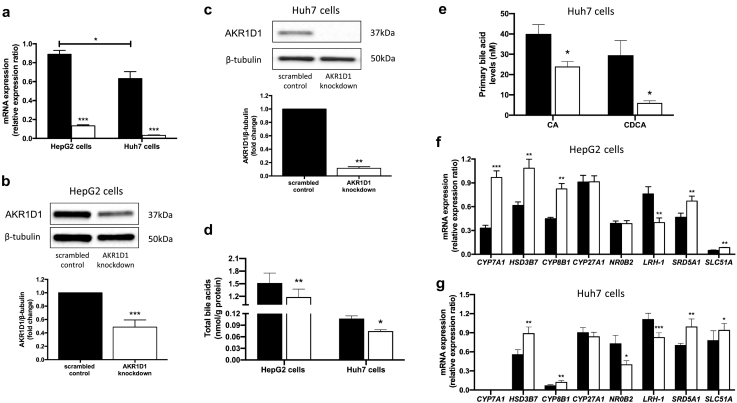
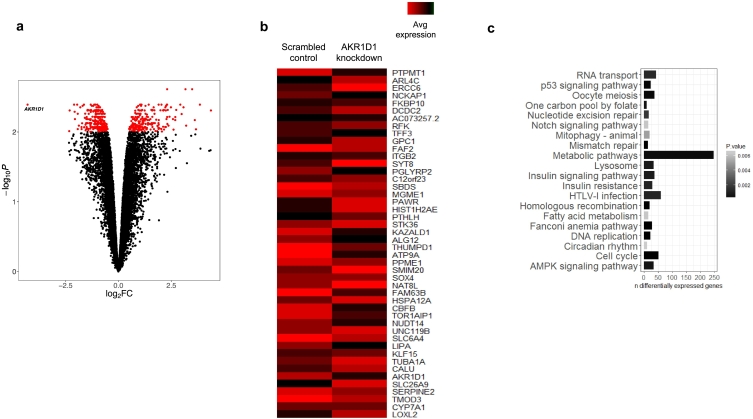
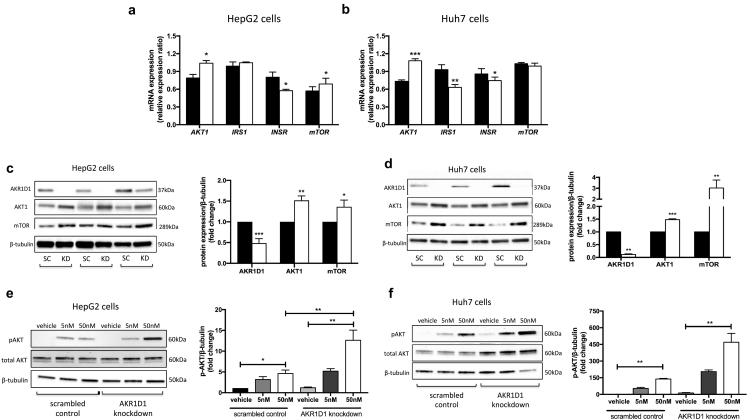
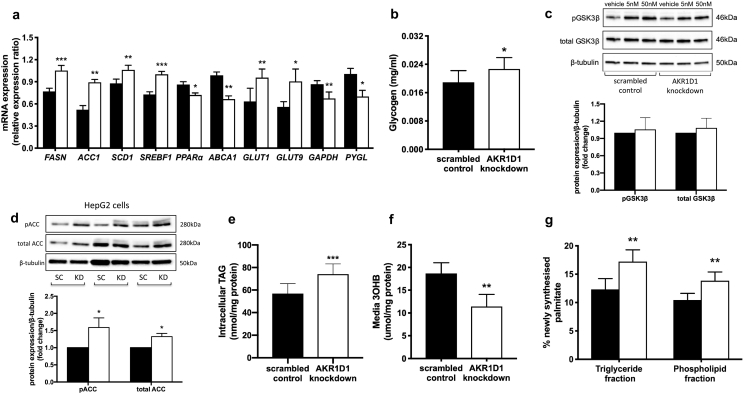
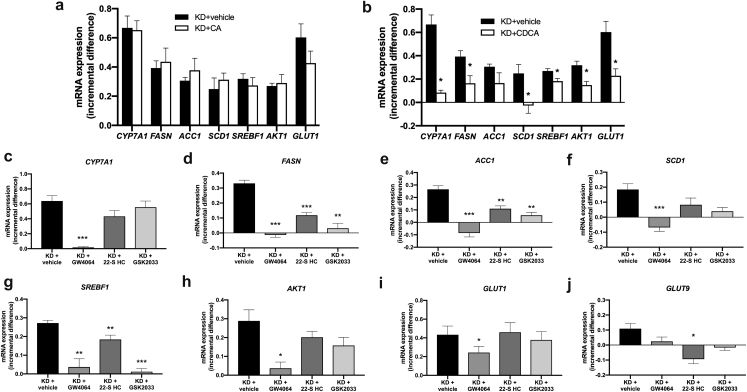
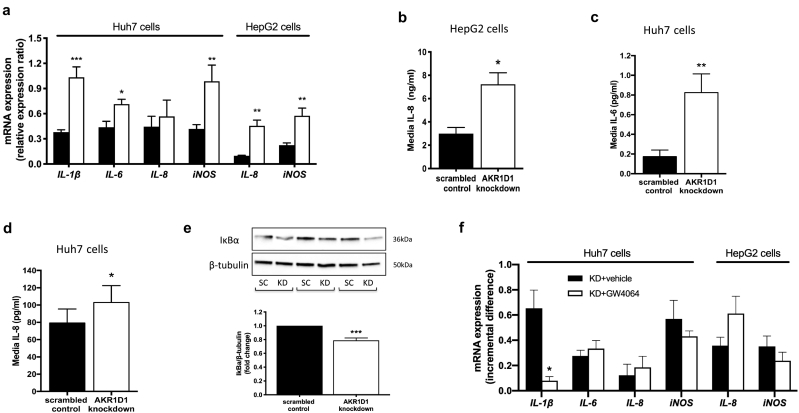
Results: In human liver biopsies, AKR1D1 expression decreased with advancing steatosis, fibrosis and inflammation. Expression was decreased in patients with type 2 diabetes. In human liver cell lines, AKR1D1 knockdown decreased primary bile acid biosynthesis and steroid hormone clearance. RNA-sequencing identified disruption of key metabolic pathways, including insulin action and fatty acid metabolism. AKR1D1 knockdown increased hepatocyte triglyceride accumulation, insulin sensitivity, and glycogen synthesis, through increased de novo lipogenesis and decreased β-oxidation, fueling hepatocyte inflammation. Pharmacological manipulation of bile acid receptor activation prevented the induction of lipogenic and carbohydrate genes, suggesting that the observed metabolic phenotype is driven through bile acid rather than steroid hormone availability.
Conclusions: Genetic manipulation of AKR1D1 regulates the metabolic phenotype of human hepatoma cell lines, driving steatosis and inflammation. Taken together, the observation that AKR1D1 mRNA is down-regulated with advancing NAFLD suggests that it may have a crucial role in the pathogenesis and progression of the disease.
Keywords: 5β-Reductase; Bile acids; Diabetes; FXR; NAFLD; Triglyceride.
Copyright © 2019 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Nothing to declare. TMP is a consultant for Research Institute for Fragrance Materials, is a recipient of a sponsored research agreement from Forendo, and is founding director of Penzymes, LLC.
Figures
References
-
- Ekstedt M., Hagström H., Nasr P., Fredrikson M., Stål P., Kechagias S. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61(5):1547–1554. - PubMed
-
- Wong R.J., Aguilar M., Cheung R., Perumpail R.B., Harrison S.A., Younossi Z.M. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology. 2015;148(3):547–555. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
