

Lipid and immune abnormalities causing age-dependent neurodegeneration and Parkinson's disease
- PMID: 31331333
- PMCID: PMC6647317
- DOI: 10.1186/s12974-019-1532-2
Lipid and immune abnormalities causing age-dependent neurodegeneration and Parkinson's disease
Abstract
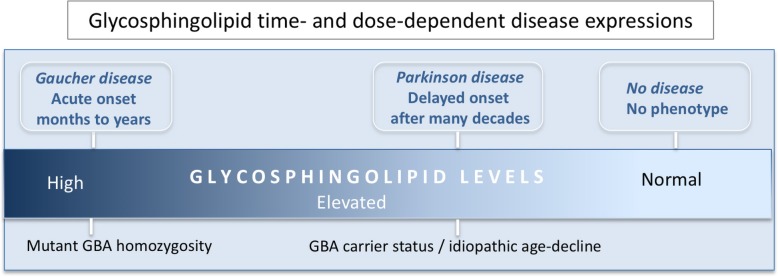
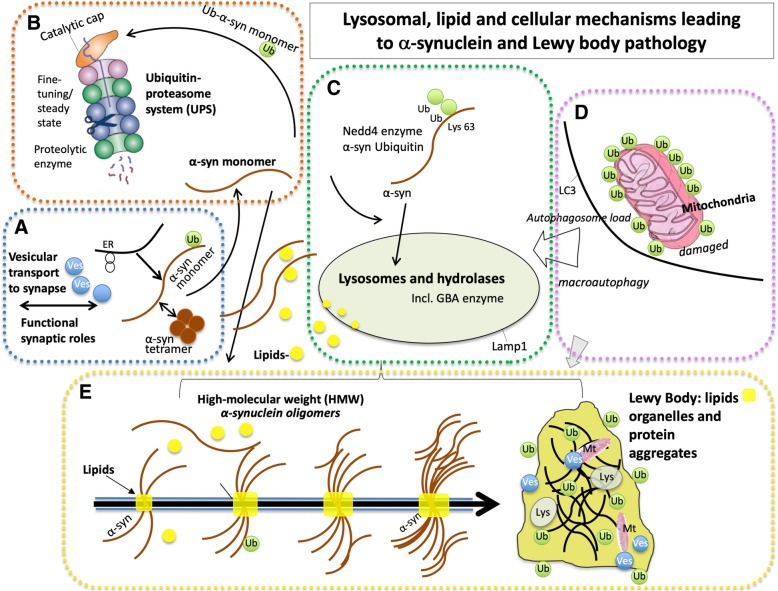
This article describes pathogenic concepts and factors, in particular glycolipid abnormalities, that create cell dysfunction and synaptic loss in neurodegenerative diseases. By phenocopying lysosomal storage disorders, such as Gaucher disease and related disorders, age- and dose-dependent changes in glycolipid cell metabolism can lead to Parkinson's disease and related dementias. Recent results show that perturbation of sphingolipid metabolism can precede or is a part of abnormal protein handling in both genetic and idiopathic Parkinson's disease and Lewy body dementia. In aging and genetic predisposition with lipid disturbance, α-synuclein's normal vesicular and synaptic role may be detrimentally shifted toward accommodating and binding such lipids. Specific neuronal glycolipid, protein, and vesicular interactions create potential pathophysiology that is amplified by astroglial and microglial immune mechanisms resulting in neurodegeneration. This perspective provides a new logic for therapeutic interventions that do not focus on protein aggregation, but rather provides a guide to the complex biology and the common sequence of events that lead to age-dependent neurodegenerative disorders.
Keywords: APOE; Apolipoprotein; Astroglia; GBA; Immune; Inflammation; Lipids; Lysosome; Microglia; Neurons; Tau; α-Synuclein.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Shi H, Belbin O, Medway C, Brown K, Kalsheker N, Carrasquillo M, Proitsi P, Powell J, Lovestone S, Goate A, et al. Genetic variants influencing human aging from late-onset Alzheimer’s disease (LOAD) genome-wide association studies (GWAS) Neurobiol Aging. 2012;33:1849.e1845–1849.e1818. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
