

Multibatch TMT Reveals False Positives, Batch Effects and Missing Values
- PMID: 31332098
- PMCID: PMC6773557
- DOI: 10.1074/mcp.RA119.001472
Multibatch TMT Reveals False Positives, Batch Effects and Missing Values
Abstract
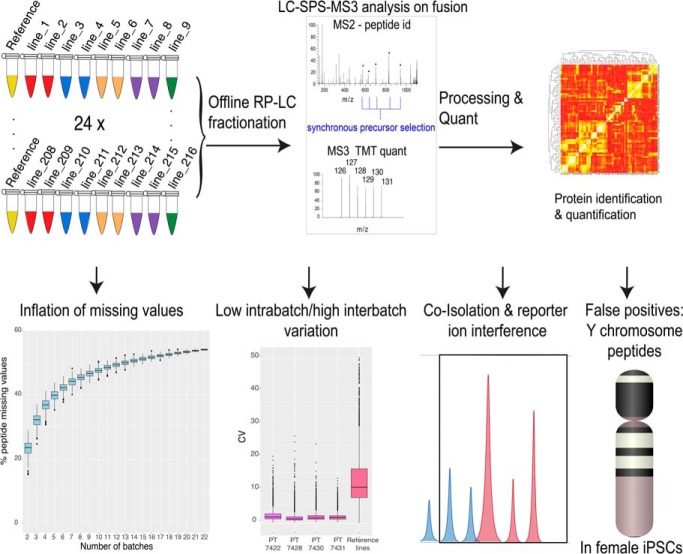
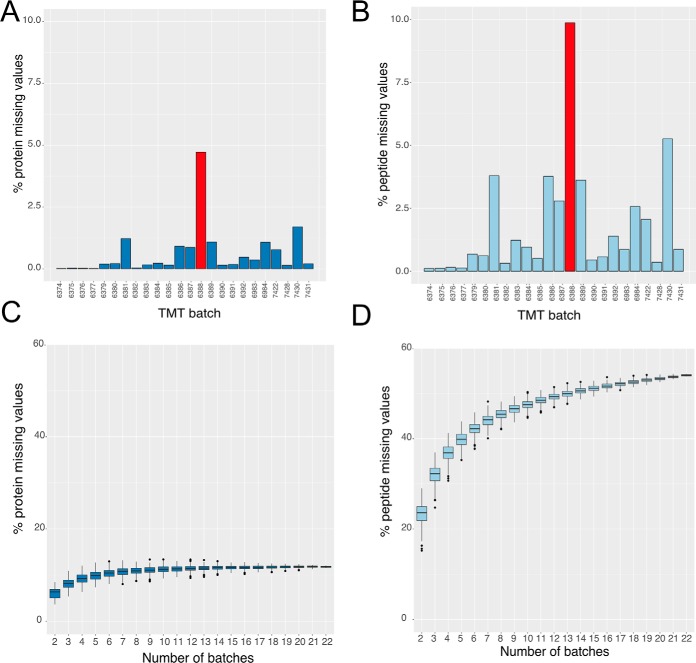
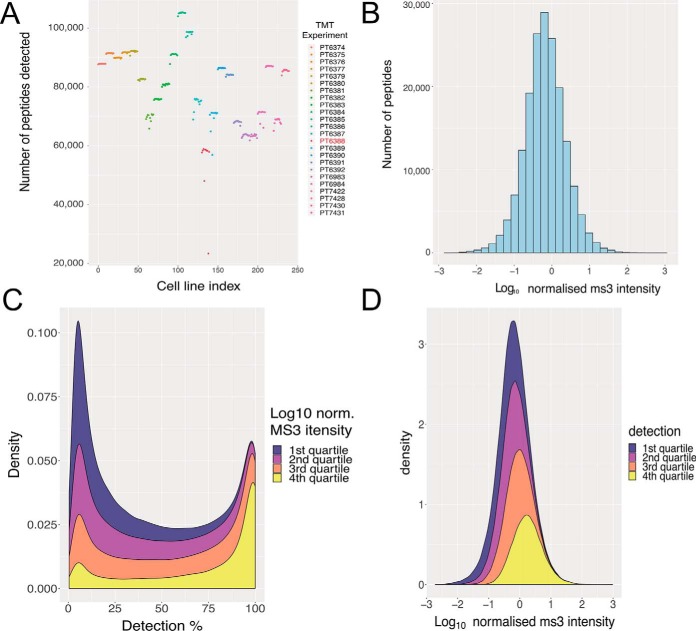
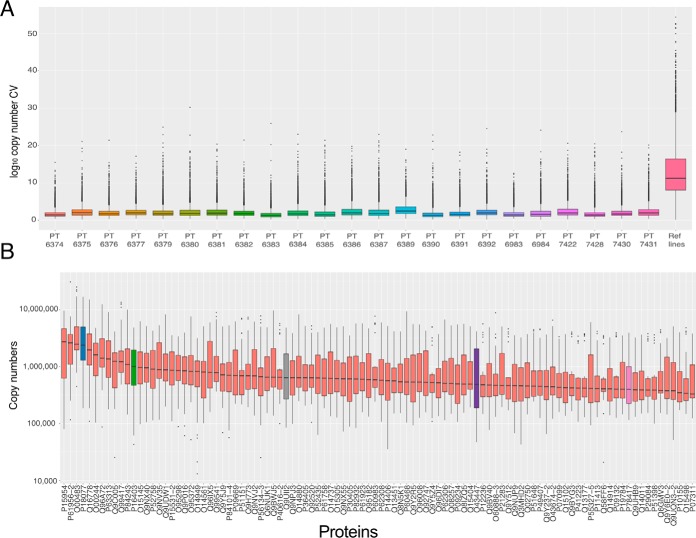
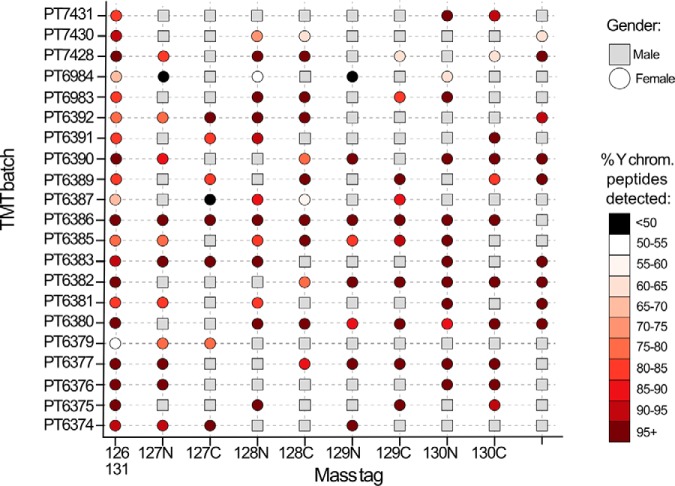
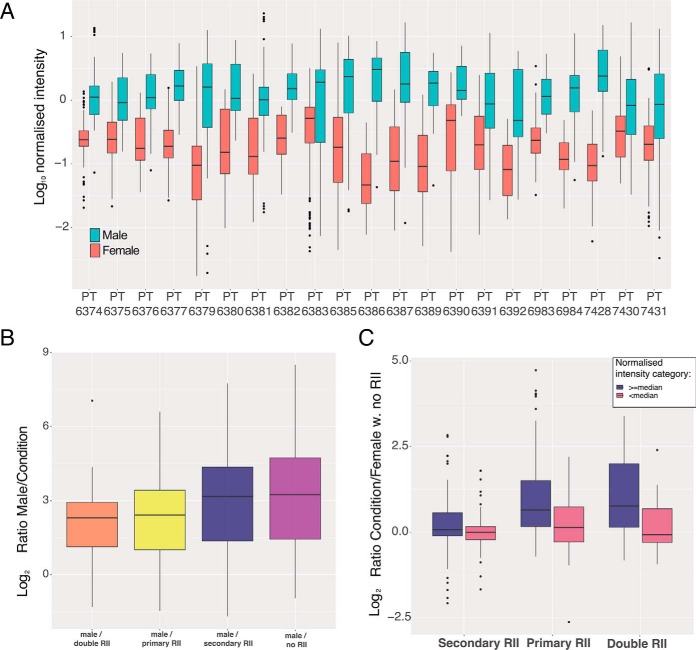
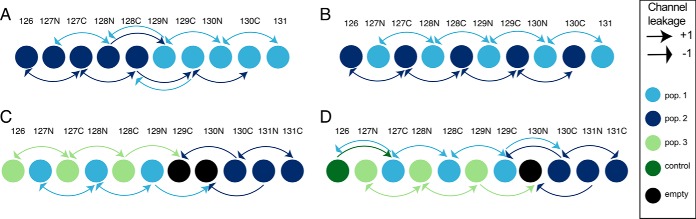
Multiplexing strategies for large-scale proteomic analyses have become increasingly prevalent, tandem mass tags (TMT) in particular. Here we used a large iPSC proteomic experiment with twenty-four 10-plex TMT batches to evaluate the effect of integrating multiple TMT batches within a single analysis. We identified a significant inflation rate of protein missing values as multiple batches are integrated and show that this pattern is aggravated at the peptide level. We also show that without normalization strategies to address the batch effects, the high precision of quantitation within a single multiplexed TMT batch is not reproduced when data from multiple TMT batches are integrated.Further, the incidence of false positives was studied by using Y chromosome peptides as an internal control. The iPSC lines quantified in this data set were derived from both male and female donors, hence the peptides mapped to the Y chromosome should be absent from female lines. Nonetheless, these Y chromosome-specific peptides were consistently detected in the female channels of all TMT batches. We then used the same Y chromosome specific peptides to quantify the level of ion coisolation as well as the effect of primary and secondary reporter ion interference. These results were used to propose solutions to mitigate the limitations of multi-batch TMT analyses. We confirm that including a common reference line in every batch increases precision by facilitating normalization across the batches and we propose experimental designs that minimize the effect of cross population reporter ion interference.
Keywords: Tandem mass spectrometry; bioinformatics; computational biology; data analysis; data evaluation; false positives; ipsc; isobaric tags; mass spectrometry.
© 2019 Brenes et al.
Figures
References
-
- Bekker-Jensen D. B., Kelstrup C. D., Batth T. S., Larsen S. C., Haldrup C., Bramsen J. B., Sorensen K. D., Hoyer S., Orntoft T. F., Andersen C. L., Nielsen M. L., and Olsen J. V. (2017) An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 4, 587–599 e584 - PMC - PubMed
-
- Meier F., Geyer P. E., Virreira Winter S., Cox J., and Mann M. (2018) BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 15, 440–448 - PubMed
-
- Camerini S. and Mauri P. (2015) The role of protein and peptide separation before mass spectrometry analysis in clinical proteomics. J. Chromatogr. A 1381, 1–12 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
