

Full-length transcript amplification and sequencing as universal method to test mRNA integrity and biallelic expression in mismatch repair genes
- PMID: 31332305
- PMCID: PMC6870986
- DOI: 10.1038/s41431-019-0472-8
Full-length transcript amplification and sequencing as universal method to test mRNA integrity and biallelic expression in mismatch repair genes
Abstract
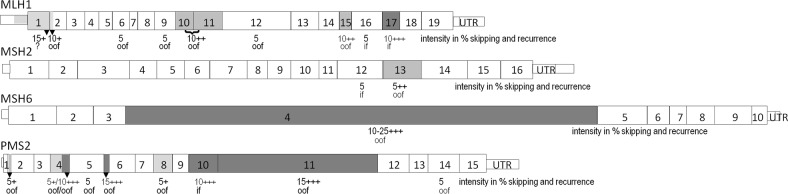
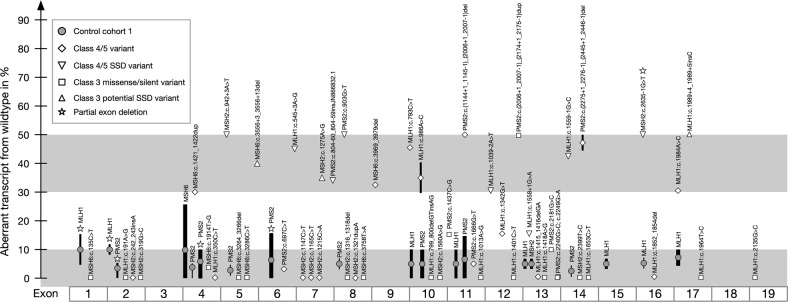
In pathogenicity assessment, RNA-based analyses are important for the correct classification of variants, and require gene-specific cut-offs for allelic representation and alternative/aberrant splicing. Beside this, the diagnostic yield of RNA-based techniques capable to detect aberrant splicing or allelic loss due to intronic/regulatory variants has to be elaborated. We established a cDNA analysis for full-length transcripts (FLT) of the four DNA mismatch repair (MMR) genes to investigate the splicing pattern and transcript integrity with active/inhibited nonsense-mediated mRNA-decay (NMD). Validation was based on results from normal controls, samples with premature termination codons (PTC), samples with splice-site defects (SSD), and samples with pathogenic putative missense variants. The method was applied to patients with variants of uncertain significance (VUS) or unexplained immunohistochemical MMR deficiency. We categorized the allelic representation into biallelic (50 ± 10%) or allelic loss (≤10%), and >10% and <40% as unclear. We defined isoforms up to 10% and exon-specific exceptions as alternative splicing, set the cut-off for SSD in cDNA + P to 30-50%, and regard >10% and <30% as unclear. FLT cDNA analyses designated 16% of all putative missense variants and 12% of VUS as SSD, detected MMR-defects in 19% of the unsolved patients, and re-classified >30% of VUS. Our method allows a standardized, systematic cDNA analysis of the MMR FLTs to assess the pathogenicity mechanism of VUS on RNA level, which will gain relevance for precision medicine and gene therapy. Diagnostic accuracy will be enhanced by detecting MMR defects in hitherto unsolved patients. The data generated will help to calibrate a high-throughput NGS-based mRNA-analysis and optimize prediction programs.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Mangold E, Pagenstecher C, Friedl W, Mathiak M, Buettner R, Engel C, et al. Spectrum and frequencies of mutations in MSH2 and MLH1 identified in 1,721 German families suspected of hereditary nonpolyposis colorectal cancer. Int J Cancer. 2005;116:692–702. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
