

Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth's biomes
- PMID: 31332386
- PMCID: PMC6813254
- DOI: 10.1038/s41564-019-0510-x
Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth's biomes
Erratum in
-
Author Correction: Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth's biomes.Nat Microbiol. 2020 Mar;5(3):527. doi: 10.1038/s41564-020-0681-5. Nat Microbiol. 2020. PMID: 32047285 Free PMC article.
Abstract
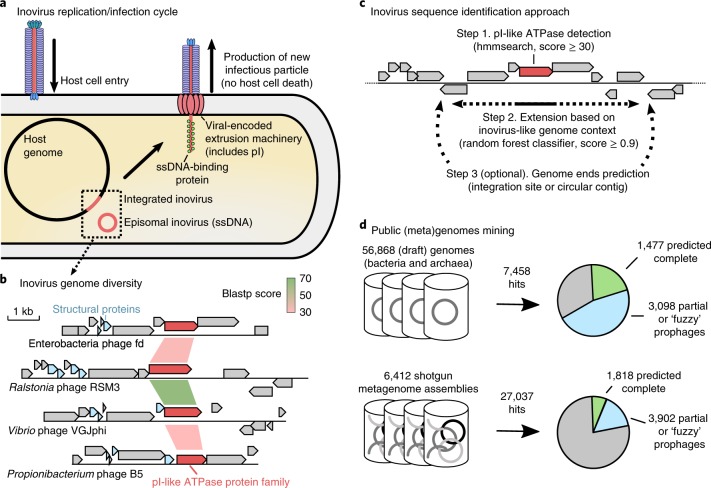
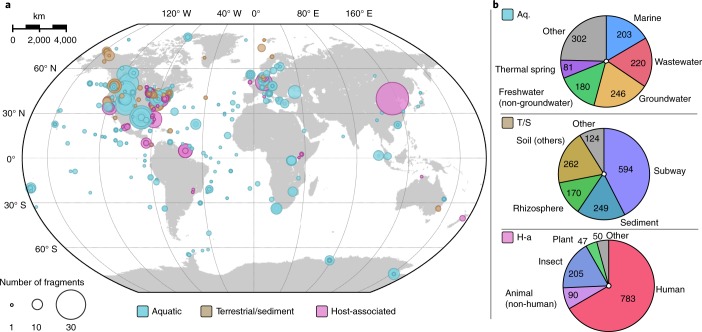
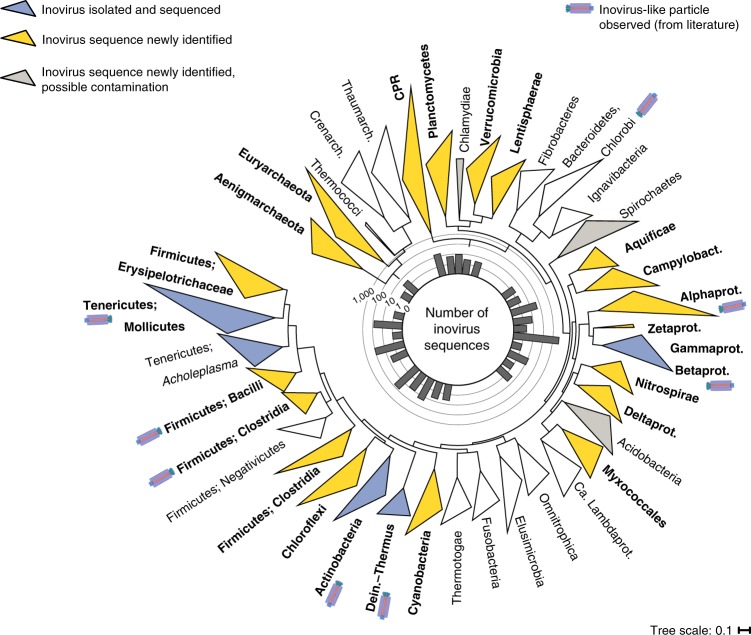
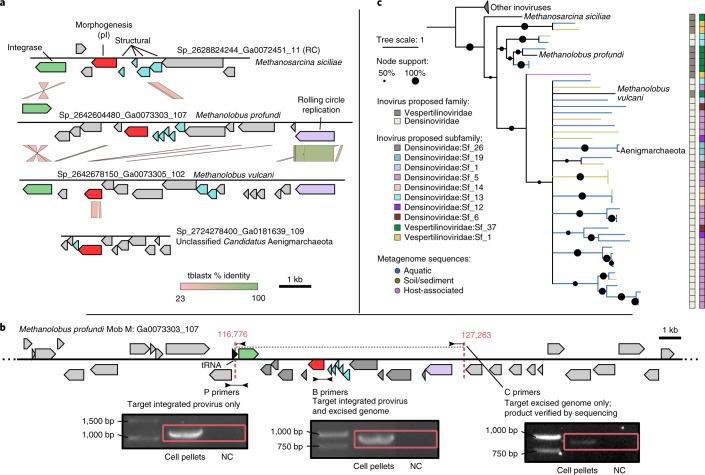
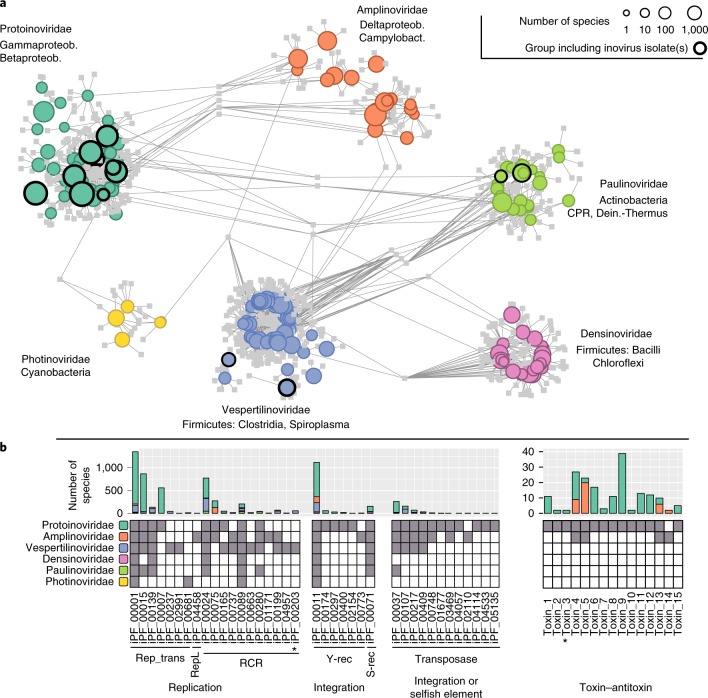
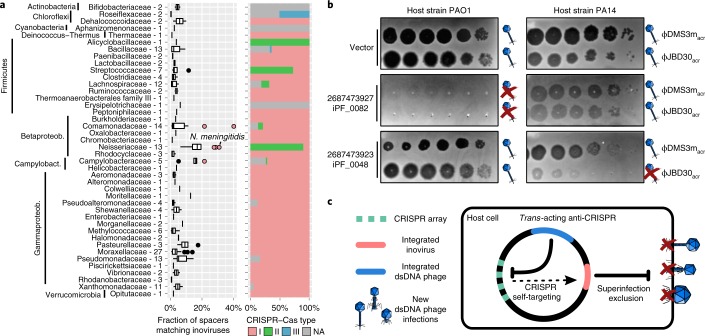
Bacteriophages from the Inoviridae family (inoviruses) are characterized by their unique morphology, genome content and infection cycle. One of the most striking features of inoviruses is their ability to establish a chronic infection whereby the viral genome resides within the cell in either an exclusively episomal state or integrated into the host chromosome and virions are continuously released without killing the host. To date, a relatively small number of inovirus isolates have been extensively studied, either for biotechnological applications, such as phage display, or because of their effect on the toxicity of known bacterial pathogens including Vibrio cholerae and Neisseria meningitidis. Here, we show that the current 56 members of the Inoviridae family represent a minute fraction of a highly diverse group of inoviruses. Using a machine learning approach leveraging a combination of marker gene and genome features, we identified 10,295 inovirus-like sequences from microbial genomes and metagenomes. Collectively, our results call for reclassification of the current Inoviridae family into a viral order including six distinct proposed families associated with nearly all bacterial phyla across virtually every ecosystem. Putative inoviruses were also detected in several archaeal genomes, suggesting that, collectively, members of this supergroup infect hosts across the domains Bacteria and Archaea. Finally, we identified an expansive diversity of inovirus-encoded toxin-antitoxin and gene expression modulation systems, alongside evidence of both synergistic (CRISPR evasion) and antagonistic (superinfection exclusion) interactions with co-infecting viruses, which we experimentally validated in a Pseudomonas model. Capturing this previously obscured component of the global virosphere may spark new avenues for microbial manipulation approaches and innovative biotechnological applications.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Rakonjac J, Bennett NJ, Spagnuolo J, Gagic D, Russel M. Filamentous bacteriophage: biology, phage display and nanotechnology applications. Curr. Issues Mol. Biol. 2011;13:51–76. - PubMed
-
- Fauquet CM. The diversity of single stranded DNA. Virus Biodivers. 2006;7:38–44. doi: 10.1080/14888386.2006.9712793. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
