

Deciphering the Composition and Functional Profile of the Microbial Communities in Chinese Moutai Liquor Starters
- PMID: 31333631
- PMCID: PMC6620787
- DOI: 10.3389/fmicb.2019.01540
Deciphering the Composition and Functional Profile of the Microbial Communities in Chinese Moutai Liquor Starters
Abstract
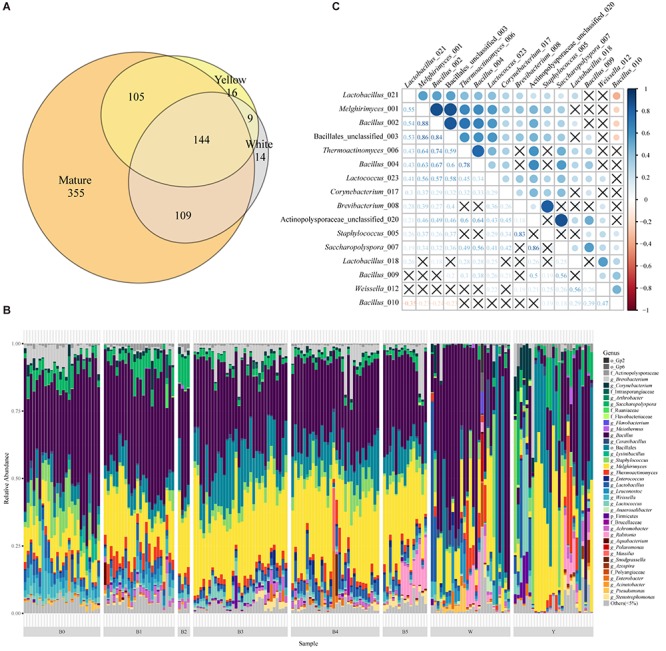
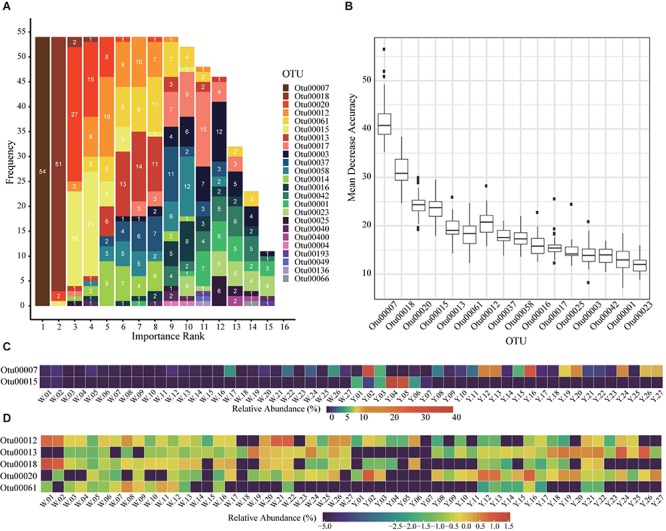
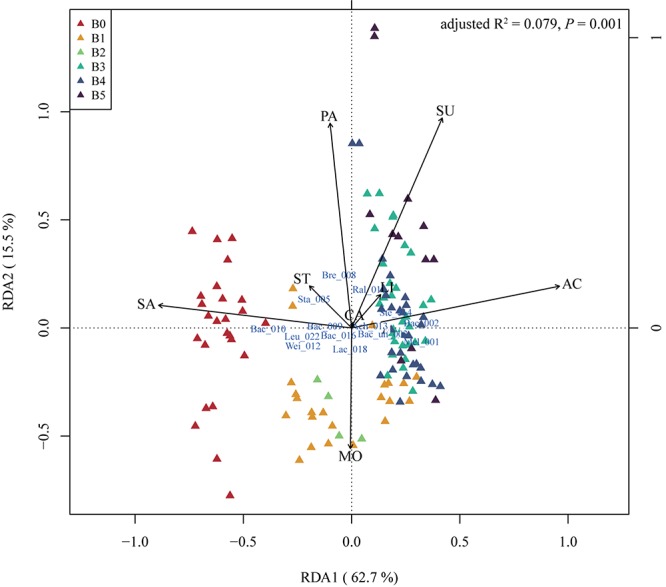
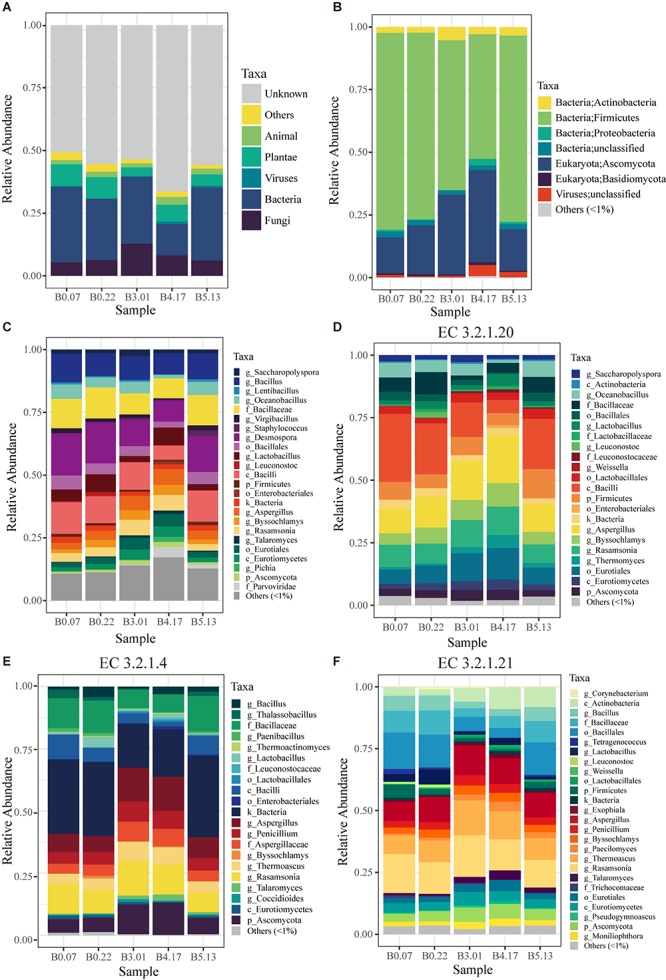
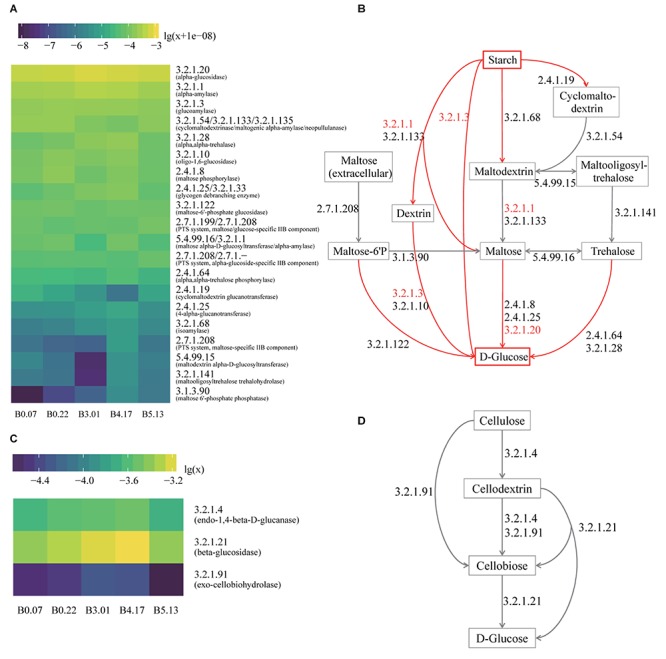
Moutai is a world-famous traditional Chinese liquor with complex taste and aroma, which are considered to be strongly influenced by the quality of fermentation starters (Daqu). However, the role of microbial communities in the starters has not been fully understood. In this study, we revealed the microbial composition of 185 Moutai starter samples, covering three different types of starters across immature and mature phases, and functional gene composition of mature starter microbiome. Our results showed that microbial composition patterns of immature starters varied, but they eventually were similar and steady when they became mature starters, after half-year storage and subsequent mixing. To help identify two types of immature starters, we selected seven operational taxonomic unit (OTU) markers by leave-one-out cross validation (LOOCV) and an OTU classified as Saccharopolyspora was the most decisive one. For mature starters, we identified a total of 16 core OTUs, one of which annotated as Bacillus was found positively associated with saccharifying power. We also identified the functional gene and microbial composition in starch and cellulose hydrolysis pathways. Microbes with higher abundances of alpha-glucosidase, alpha-amylase, and glucoamylase probably contributed to high saccharifying power. Overall, this study reveals the features of Moutai starter microbial communities in different phases and improves understanding of the relationships between microbiota and functional properties of the starters.
Keywords: 16S rRNA gene; Daqu; Moutai liquor; OTU; fermentation starter; immature starter; metagenomics; saccharifying power.
Figures
References
LinkOut - more resources
Full Text Sources