

Chromatin Profiling of the Repetitive and Nonrepetitive Genomes of the Human Fungal Pathogen Candida albicans
- PMID: 31337722
- PMCID: PMC6650553
- DOI: 10.1128/mBio.01376-19
Chromatin Profiling of the Repetitive and Nonrepetitive Genomes of the Human Fungal Pathogen Candida albicans
Abstract
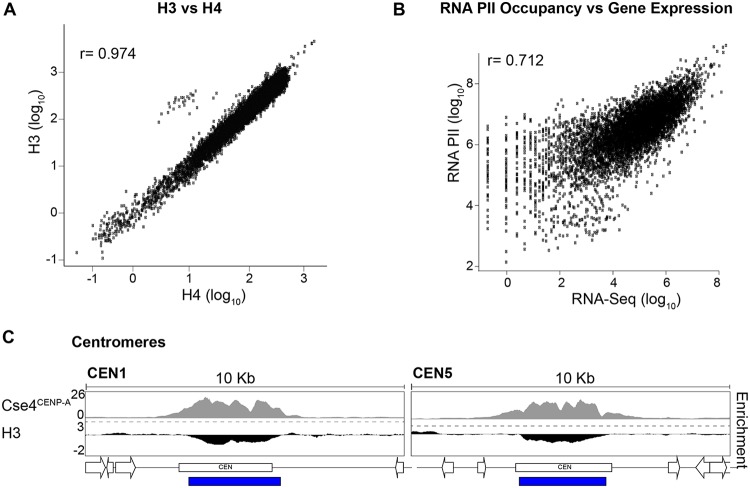
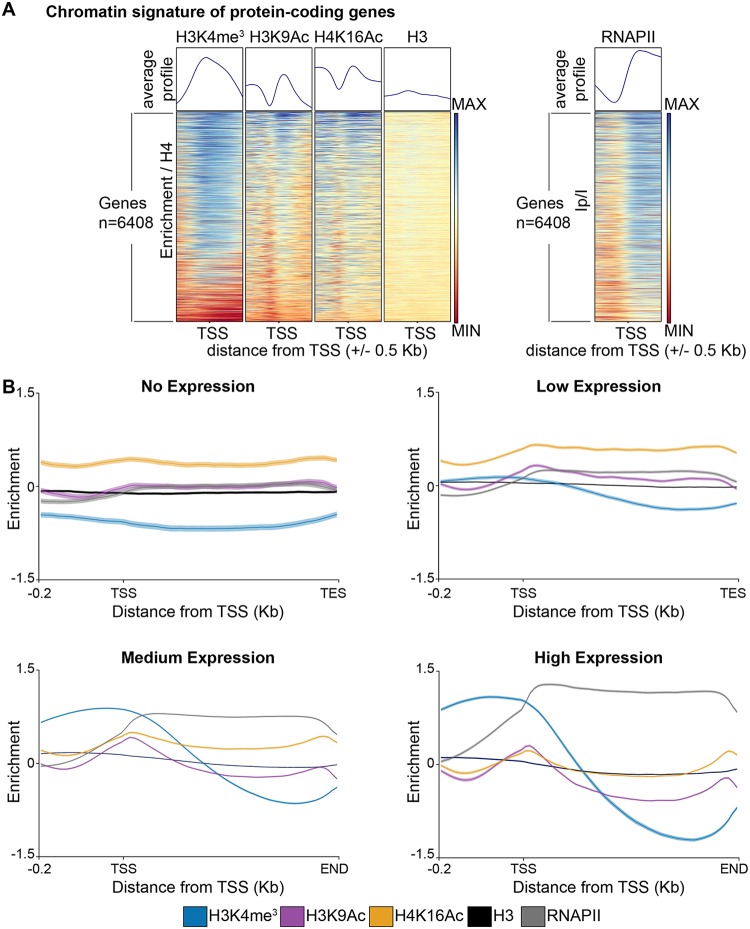
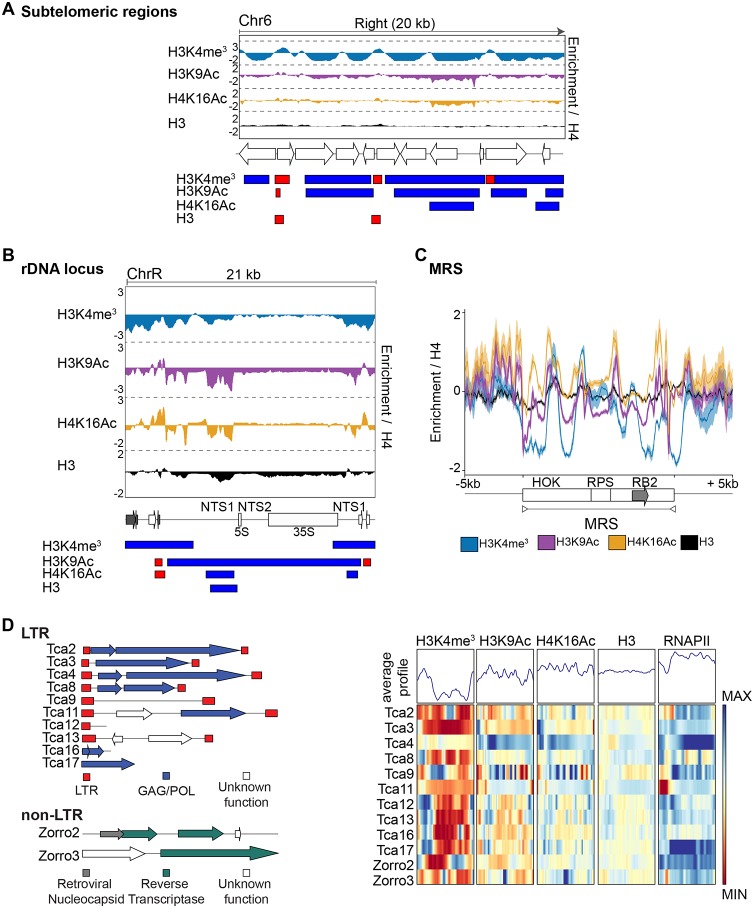
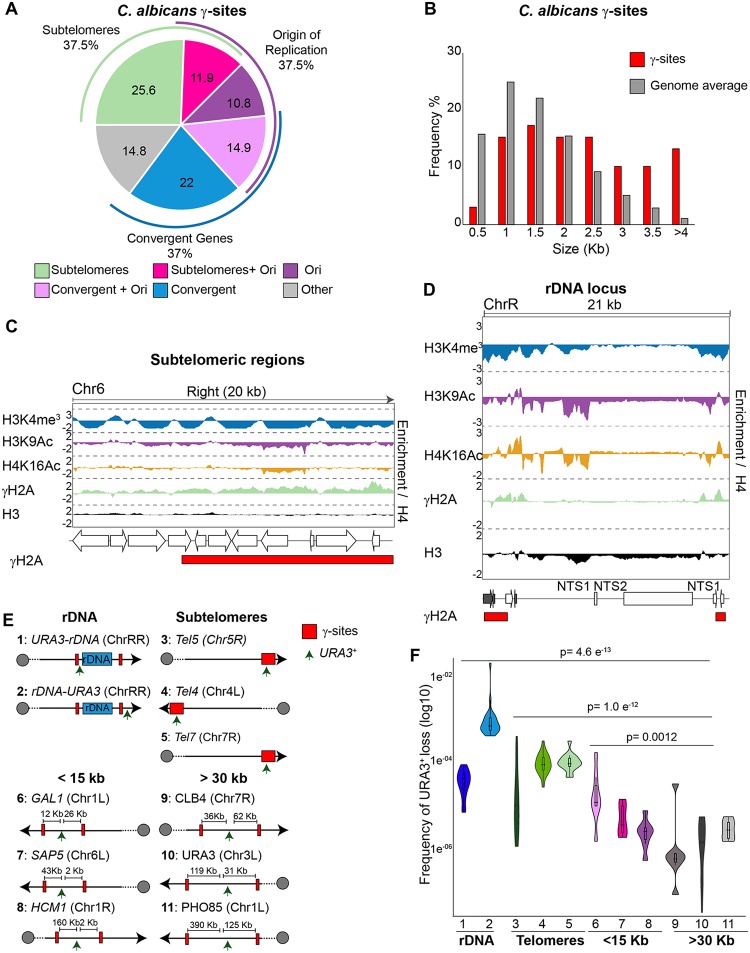
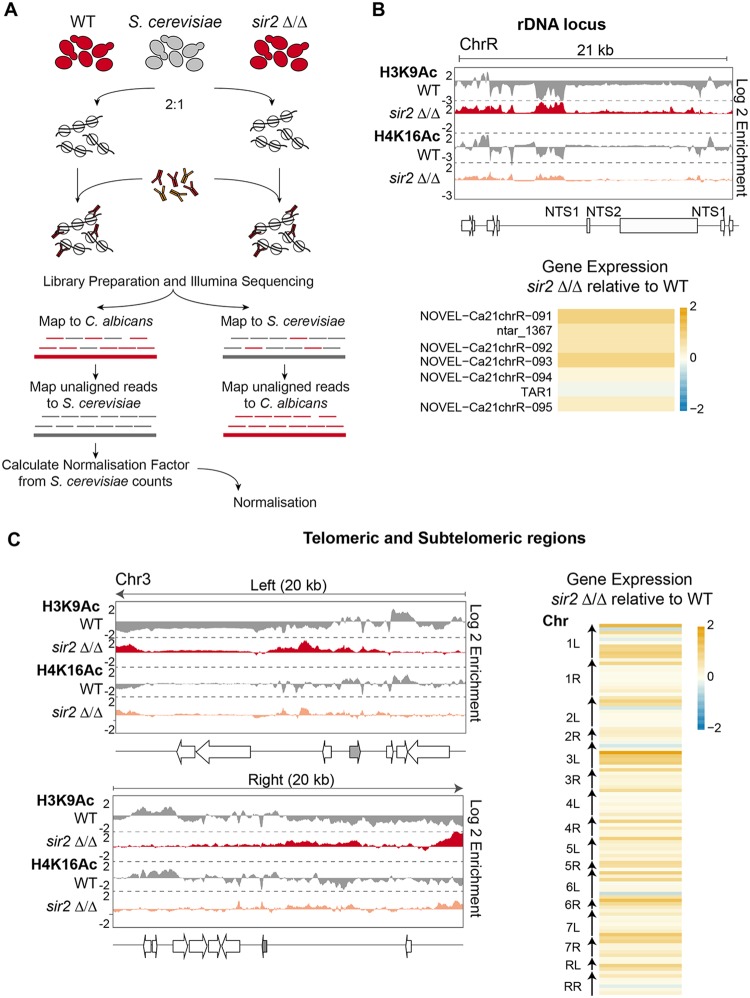
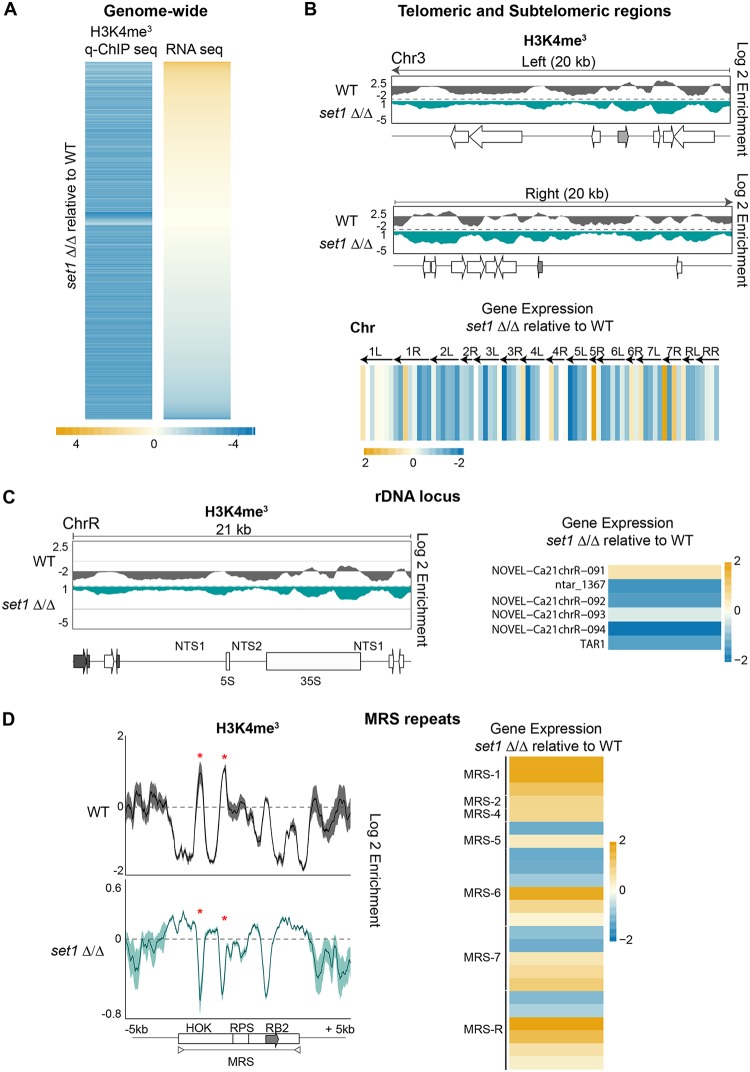
Eukaryotic genomes are packaged into chromatin structures that play pivotal roles in regulating all DNA-associated processes. Histone posttranslational modifications modulate chromatin structure and function, leading to rapid regulation of gene expression and genome stability, key steps in environmental adaptation. Candida albicans, a prevalent fungal pathogen in humans, can rapidly adapt and thrive in diverse host niches. The contribution of chromatin to C. albicans biology is largely unexplored. Here, we generated the first comprehensive chromatin profile of histone modifications (histone H3 trimethylated on lysine 4 [H3K4me3], histone H3 acetylated on lysine 9 [H3K9Ac], acetylated lysine 16 on histone H4 [H4K16Ac], and γH2A) across the C. albicans genome and investigated its relationship to gene expression by harnessing genome-wide sequencing approaches. We demonstrated that gene-rich nonrepetitive regions are packaged into canonical euchromatin in association with histone modifications that mirror their transcriptional activity. In contrast, repetitive regions are assembled into distinct chromatin states; subtelomeric regions and the ribosomal DNA (rDNA) locus are assembled into heterochromatin, while major repeat sequences and transposons are packaged in chromatin that bears features of euchromatin and heterochromatin. Genome-wide mapping of γH2A, a marker of genome instability, identified potential recombination-prone genomic loci. Finally, we present the first quantitative chromatin profiling in C. albicans to delineate the role of the chromatin modifiers Sir2 and Set1 in controlling chromatin structure and gene expression. This report presents the first genome-wide chromatin profiling of histone modifications associated with the C. albicans genome. These epigenomic maps provide an invaluable resource to understand the contribution of chromatin to C. albicans biology and identify aspects of C. albicans chromatin organization that differ from that of other yeasts.IMPORTANCE The fungus Candida albicans is an opportunistic pathogen that normally lives on the human body without causing any harm. However, C. albicans is also a dangerous pathogen responsible for millions of infections annually. C. albicans is such a successful pathogen because it can adapt to and thrive in different environments. Chemical modifications of chromatin, the structure that packages DNA into cells, can allow environmental adaptation by regulating gene expression and genome organization. Surprisingly, the contribution of chromatin modification to C. albicans biology is still largely unknown. For the first time, we analyzed C. albicans chromatin modifications on a genome-wide basis. We demonstrate that specific chromatin states are associated with distinct regions of the C. albicans genome and identify the roles of the chromatin modifiers Sir2 and Set1 in shaping C. albicans chromatin and gene expression.
Keywords: Candida albicans; chromatin; epigenetics; euchromatin; genome instability; heterochromatin.
Copyright © 2019 Price et al.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
