

Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from the North American Consortium for Histiocytosis (NACHO)
- PMID: 31339233
- PMCID: PMC7340087
- DOI: 10.1002/pbc.27929
Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from the North American Consortium for Histiocytosis (NACHO)
Abstract
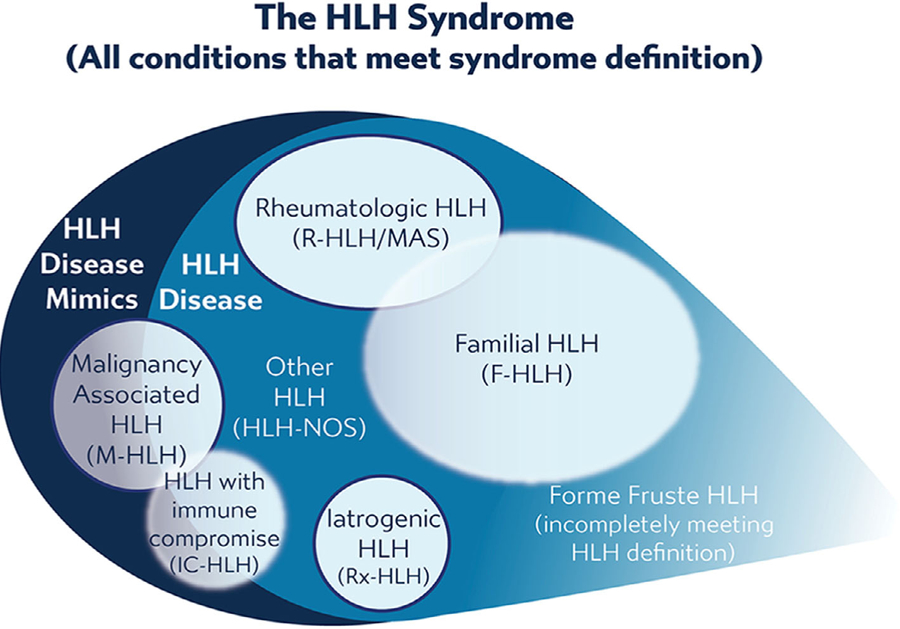
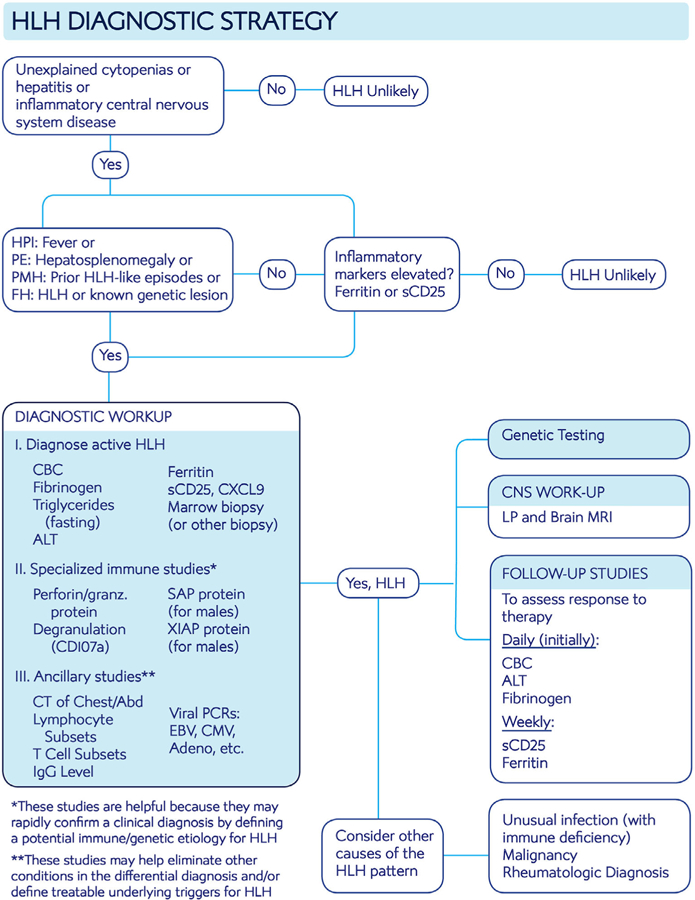
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of pathologic immune activation, often associated with genetic defects of lymphocyte cytotoxicity. Though a distinctive constellation of features has been described for HLH, diagnosis remains challenging as patients have diverse presentations associated with a variety of triggers. We propose two concepts to clarify how HLH is diagnosed and treated: within the broader syndrome of HLH, "HLH disease" should be distinguished from "HLH disease mimics" and HLH subtypes should be categorized by specific etiologic associations, not the ambiguous dichotomy of "primary" and "secondary." We provide expert-based advice regarding the diagnosis and initiation of treatment for patients with HLH, rooted in improved understanding of its pathophysiology.
Keywords: hematology; hemophagocytic lymphohistiocytosis; immunology.
© 2019 Wiley Periodicals, Inc.
Conflict of interest statement
CONFLICT OF INTEREST
Michael B. Jordan is a consultant and advisory board member for Novimmune, Sobi. Kim E. Nichols received research funding from Incyte. Ashish Kumar is a consultant for Novimmune, Sobi. Michael Henry is a consultant for Sobi. Michelle L. Hermiston is an external advisory board member for Novartis. Carl E. Allen is a consultant and advisory board member for Novimmune, Sobi.
Figures
References
-
- Ladisch S, Poplack DG, Holiman B, Blaese RM. Immunodeficiency in familial erythrophagocytic lymphohistiocytosis. Lancet. 1978;1(8064):581–583. - PubMed
-
- Jaffe ES, Costa J, Fauci AS, Cossman J, Tsokos M. Malignant lymphoma and erythrophagocytosis simulating malignant histiocytosis. Am J Med. 1983;75(5):741–749. - PubMed
-
- Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr. 1985;106(4):561–566. - PubMed
-
- Goldberg J, Nezelof C. Lymphohistiocytosis: a multi-factorial syndrome of macrophagic activation clinico-pathological study of 38 cases. Hematol Oncol. 1986;4(4):275–289. - PubMed
