

CTCF cooperates with CtIP to drive homologous recombination repair of double-strand breaks
- PMID: 31340001
- PMCID: PMC6753481
- DOI: 10.1093/nar/gkz639
CTCF cooperates with CtIP to drive homologous recombination repair of double-strand breaks
Abstract
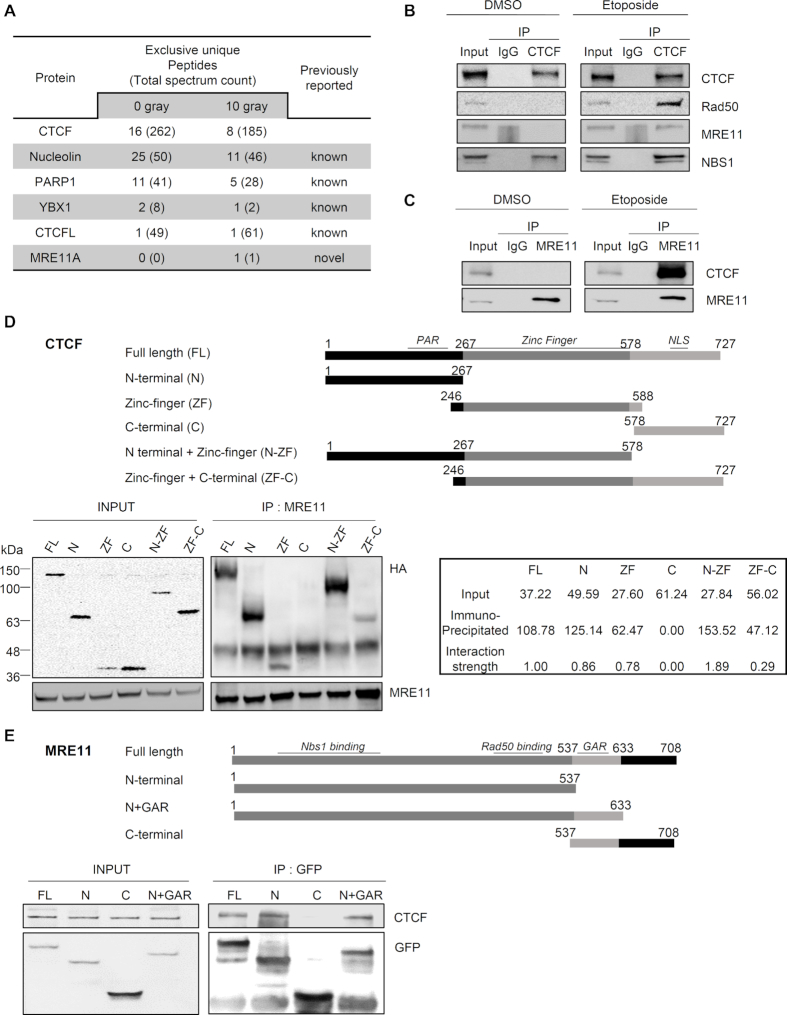
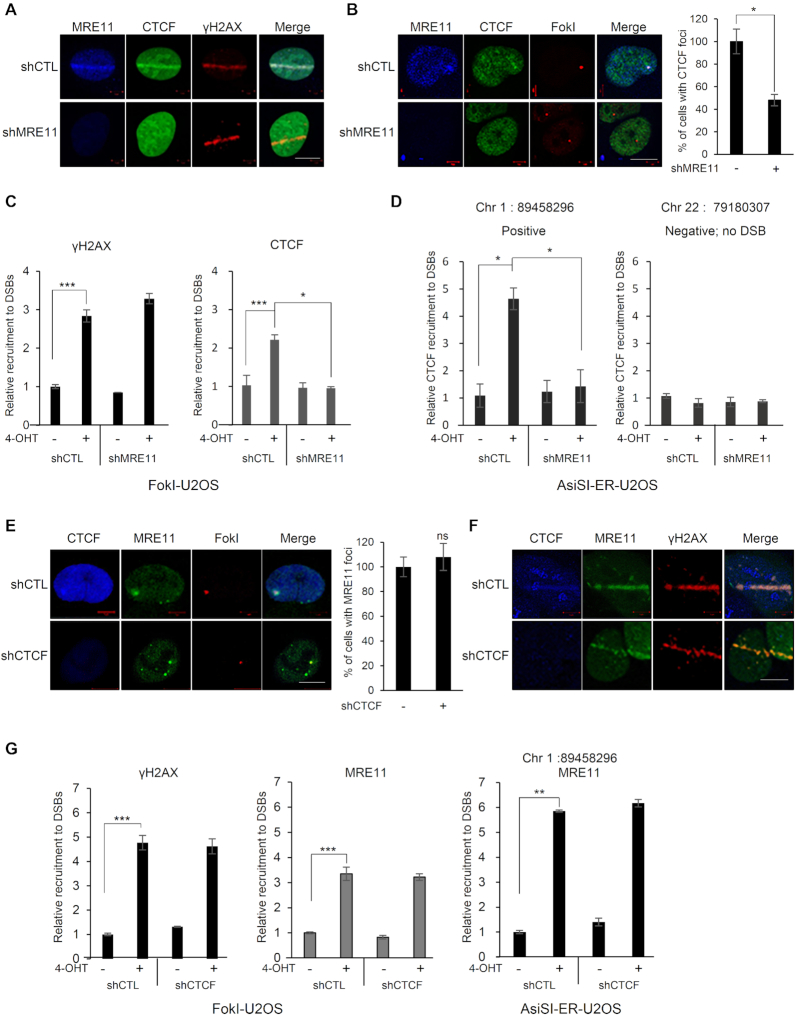
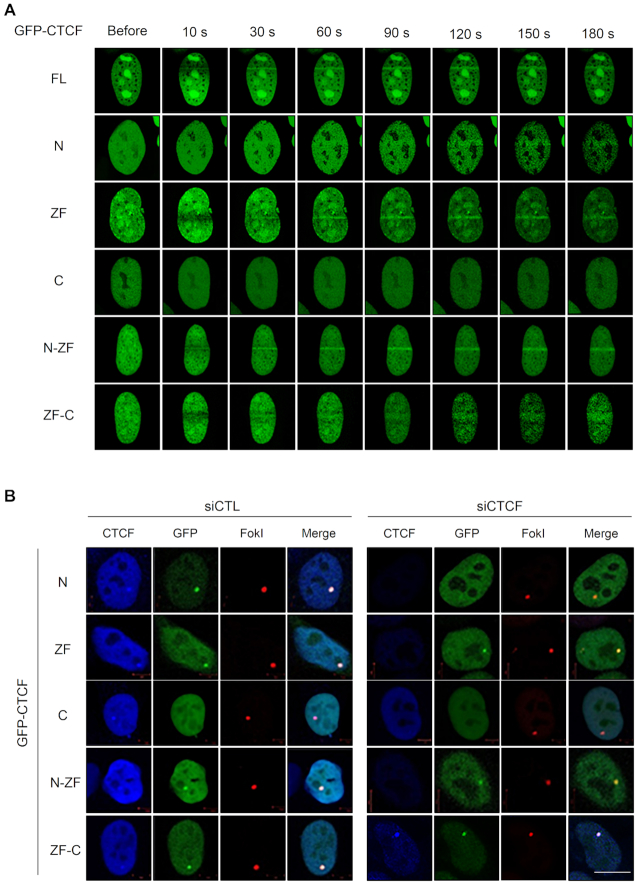
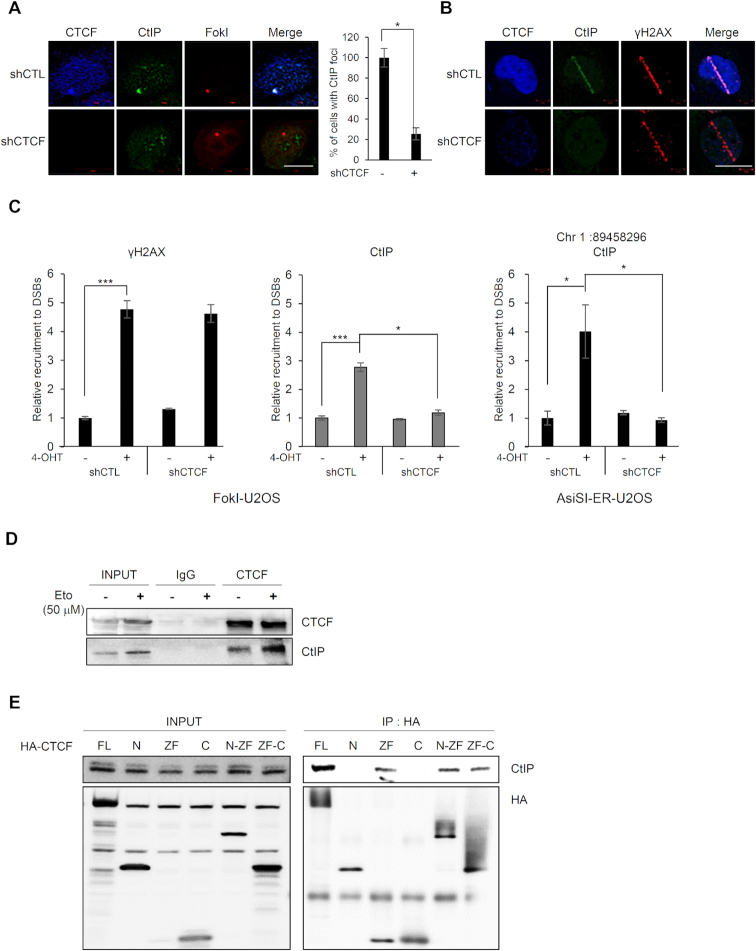
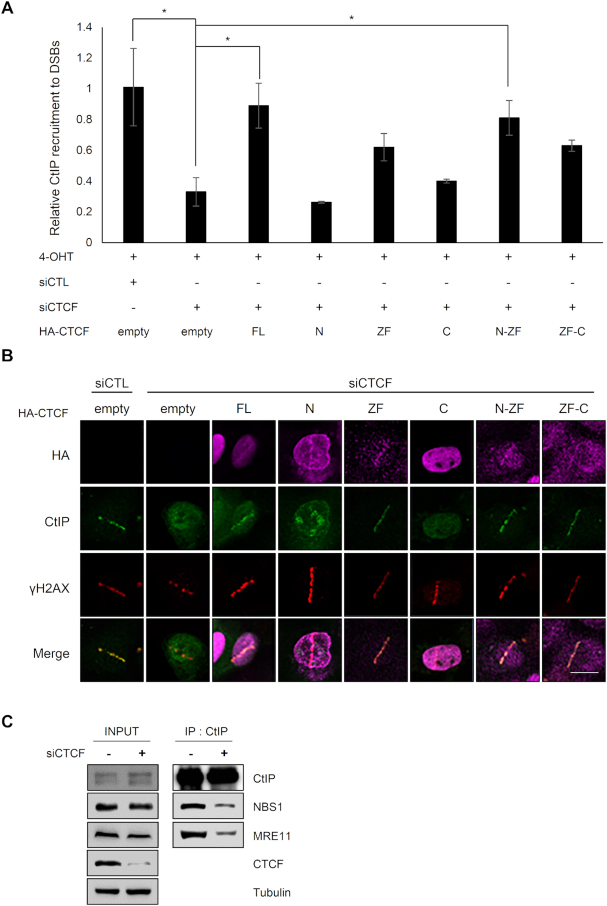
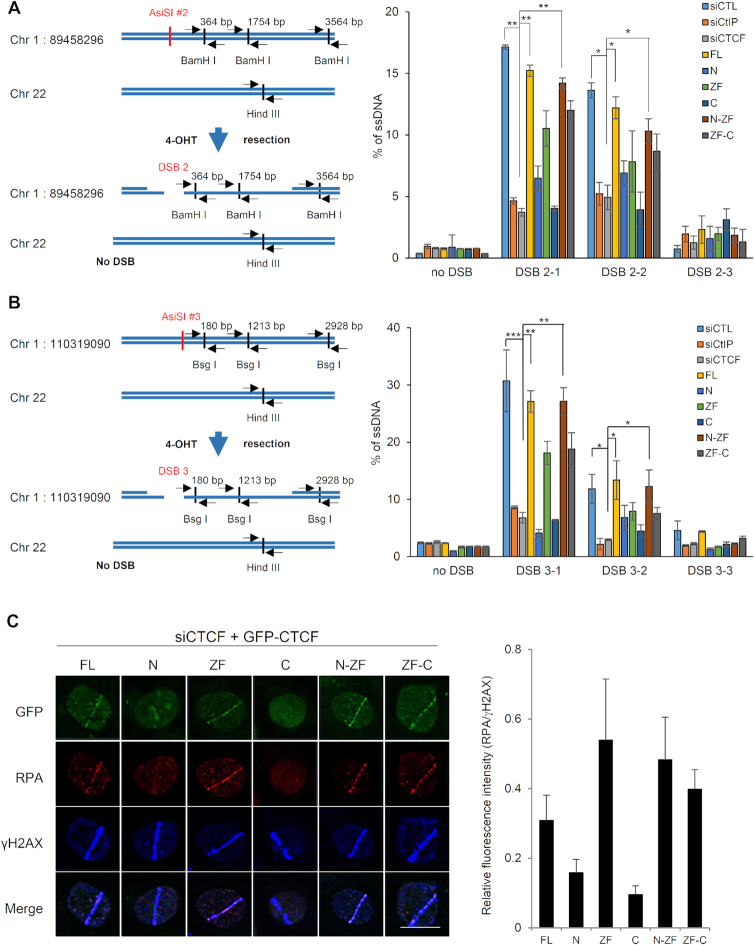
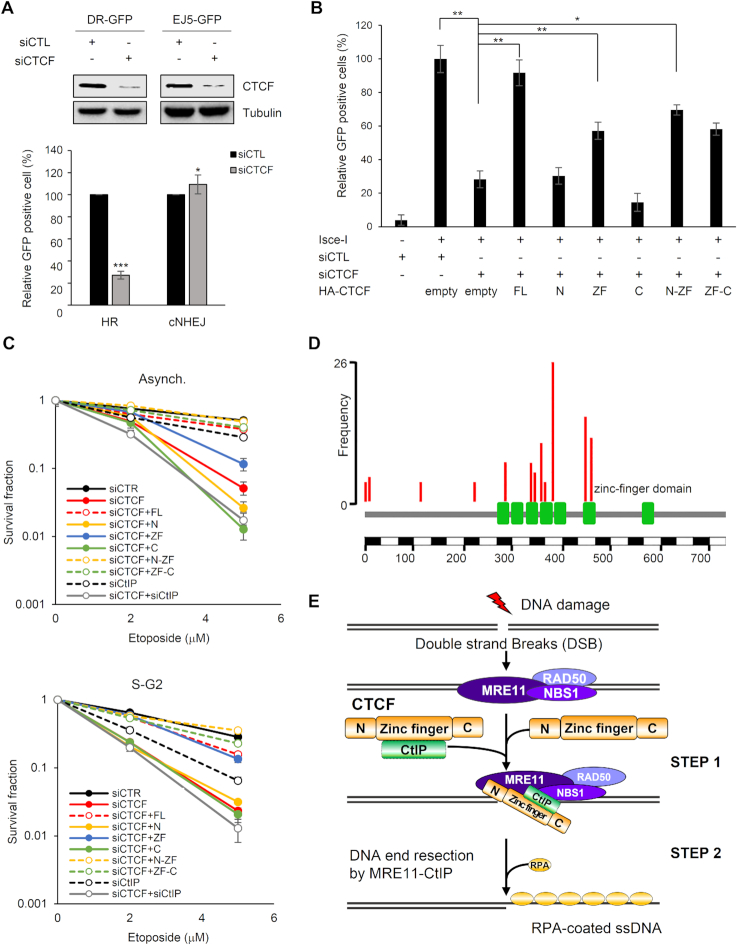
The pleiotropic CCCTC-binding factor (CTCF) plays a role in homologous recombination (HR) repair of DNA double-strand breaks (DSBs). However, the precise mechanistic role of CTCF in HR remains largely unclear. Here, we show that CTCF engages in DNA end resection, which is the initial, crucial step in HR, through its interactions with MRE11 and CtIP. Depletion of CTCF profoundly impairs HR and attenuates CtIP recruitment at DSBs. CTCF physically interacts with MRE11 and CtIP and promotes CtIP recruitment to sites of DNA damage. Subsequently, CTCF facilitates DNA end resection to allow HR, in conjunction with MRE11-CtIP. Notably, the zinc finger domain of CTCF binds to both MRE11 and CtIP and enables proficient CtIP recruitment, DNA end resection and HR. The N-terminus of CTCF is able to bind to only MRE11 and its C-terminus is incapable of binding to MRE11 and CtIP, thereby resulting in compromised CtIP recruitment, DSB resection and HR. Overall, this suggests an important function of CTCF in DNA end resection through the recruitment of CtIP at DSBs. Collectively, our findings identify a critical role of CTCF at the first control point in selecting the HR repair pathway.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- San Filippo J., Sung P., Klein H.. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008; 77:229–257. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
