

Resolving medulloblastoma cellular architecture by single-cell genomics
- PMID: 31341285
- PMCID: PMC6754173
- DOI: 10.1038/s41586-019-1434-6
Resolving medulloblastoma cellular architecture by single-cell genomics
Abstract
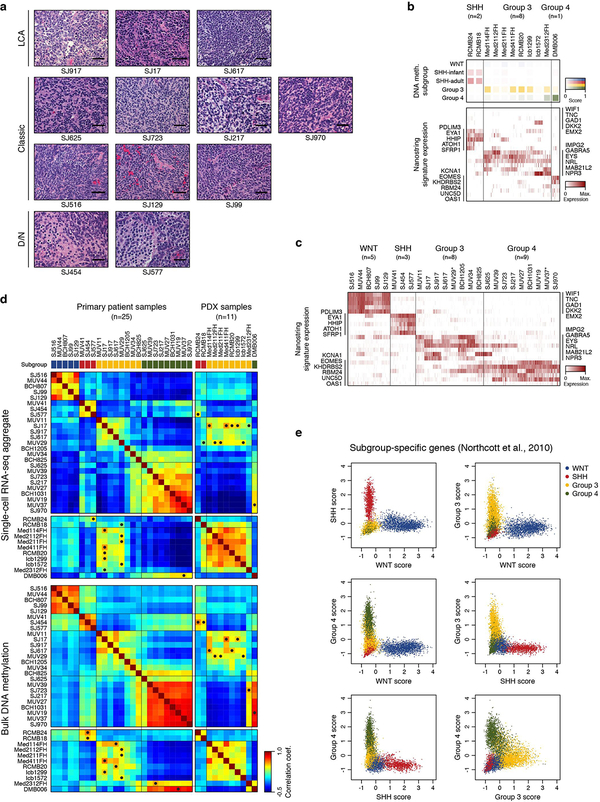
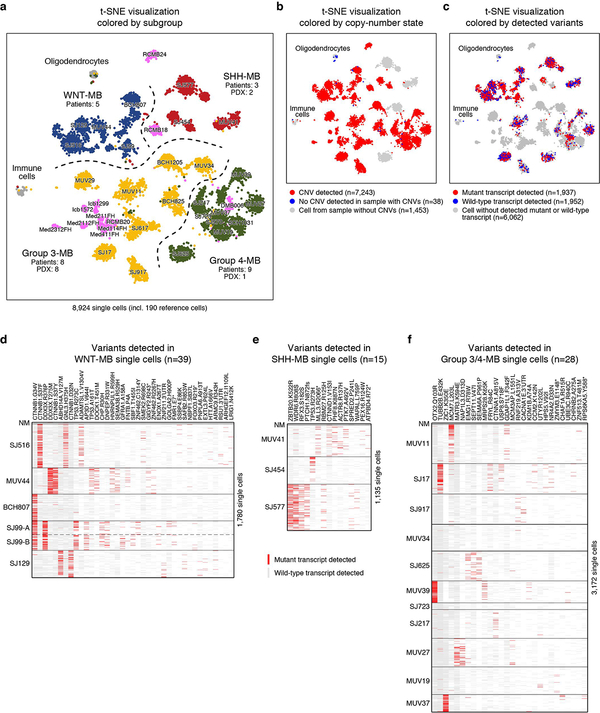
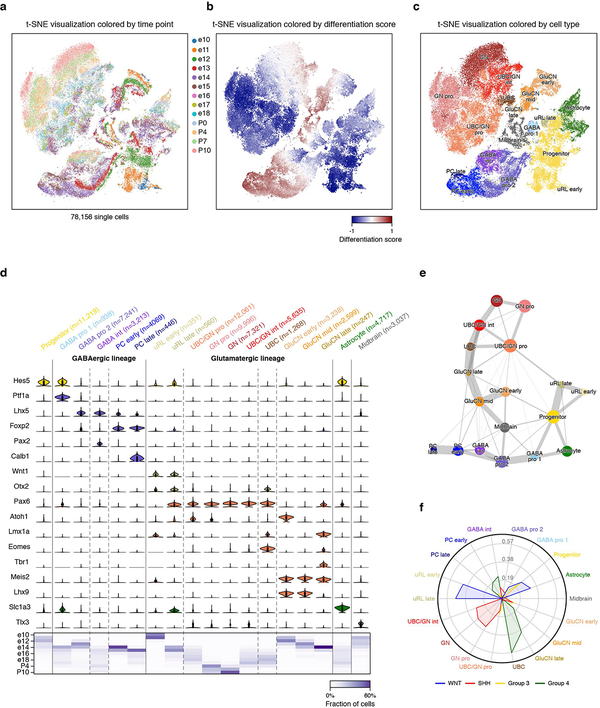
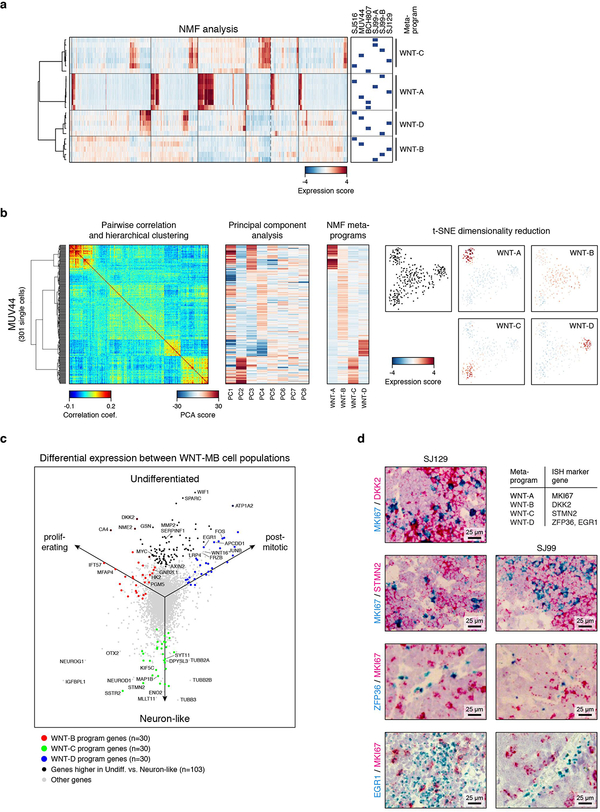
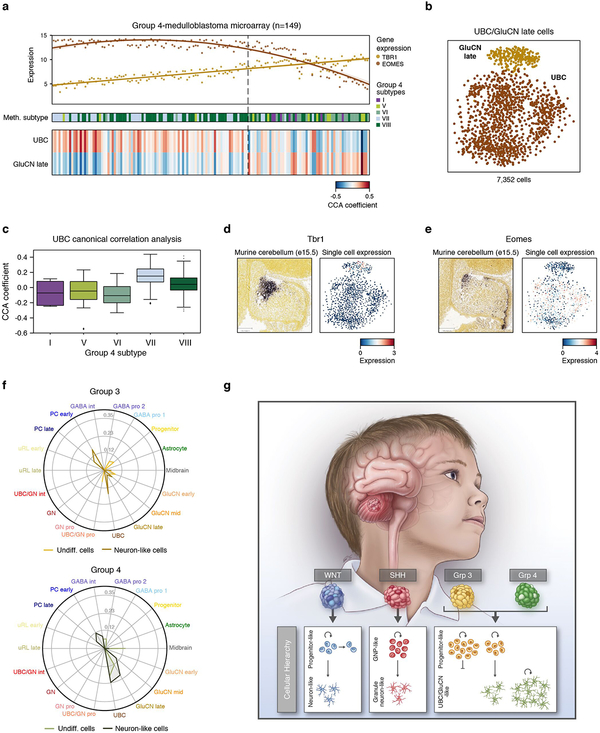
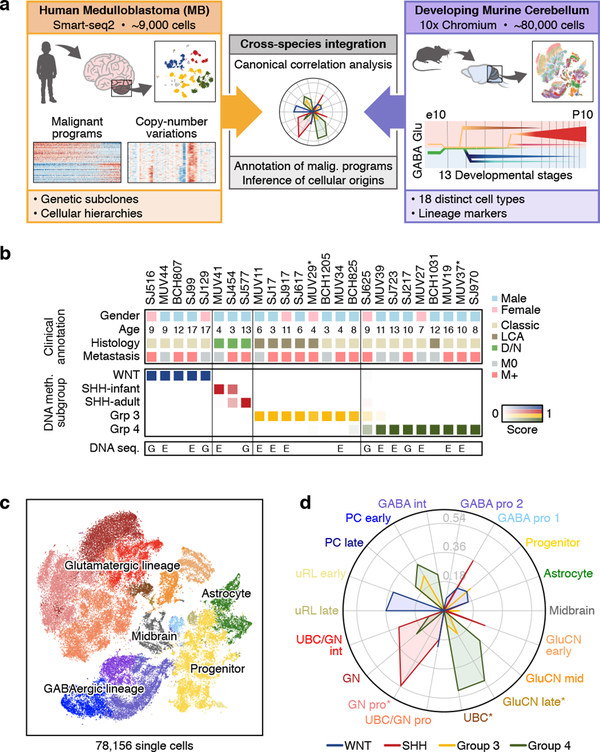
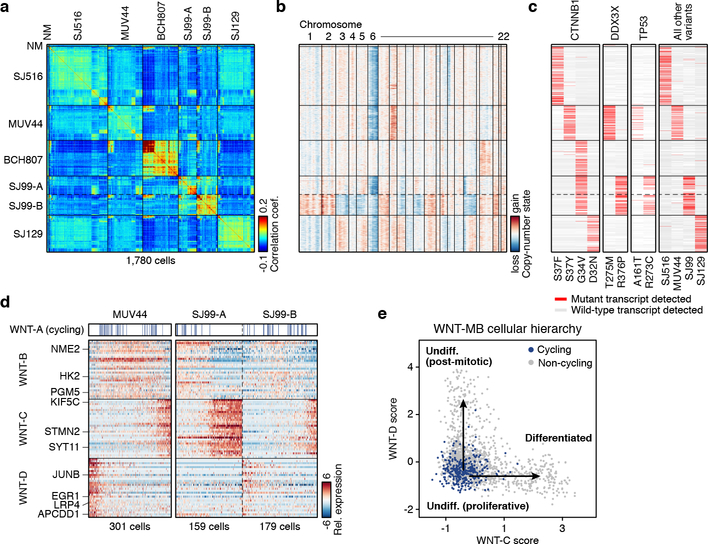
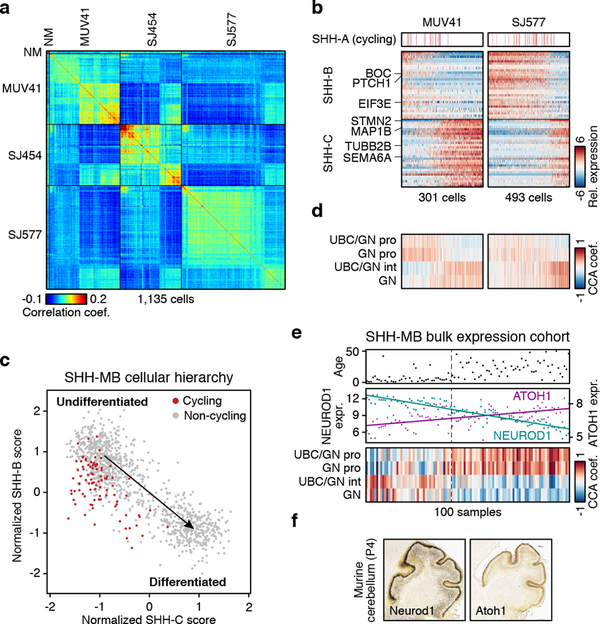
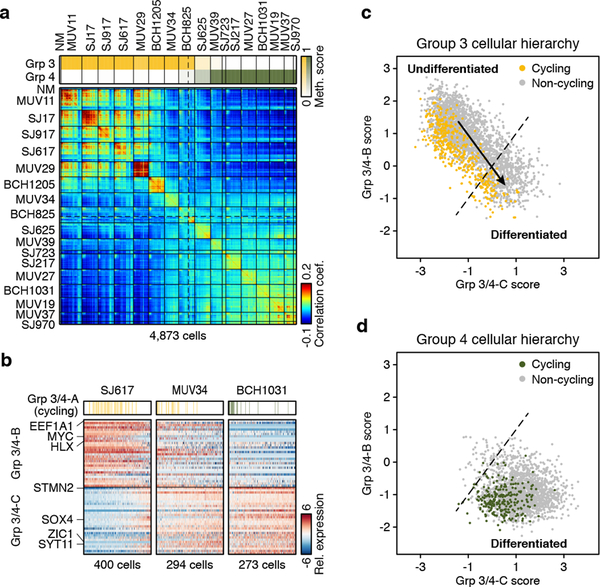
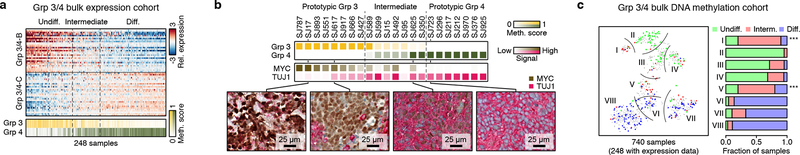
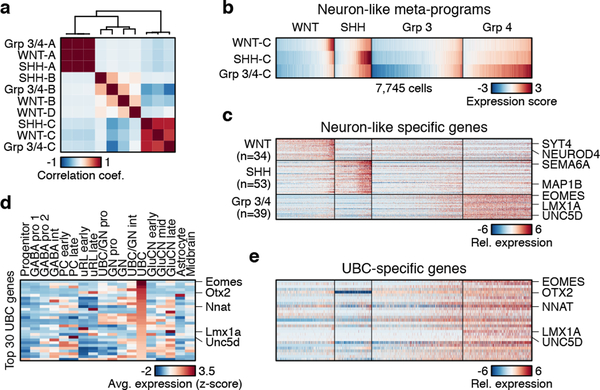
Medulloblastoma is a malignant childhood cerebellar tumour type that comprises distinct molecular subgroups. Whereas genomic characteristics of these subgroups are well defined, the extent to which cellular diversity underlies their divergent biology and clinical behaviour remains largely unexplored. Here we used single-cell transcriptomics to investigate intra- and intertumoral heterogeneity in 25 medulloblastomas spanning all molecular subgroups. WNT, SHH and Group 3 tumours comprised subgroup-specific undifferentiated and differentiated neuronal-like malignant populations, whereas Group 4 tumours consisted exclusively of differentiated neuronal-like neoplastic cells. SHH tumours closely resembled granule neurons of varying differentiation states that correlated with patient age. Group 3 and Group 4 tumours exhibited a developmental trajectory from primitive progenitor-like to more mature neuronal-like cells, the relative proportions of which distinguished these subgroups. Cross-species transcriptomics defined distinct glutamatergic populations as putative cells-of-origin for SHH and Group 4 subtypes. Collectively, these data provide insights into the cellular and developmental states underlying subtype-specific medulloblastoma biology.
Figures

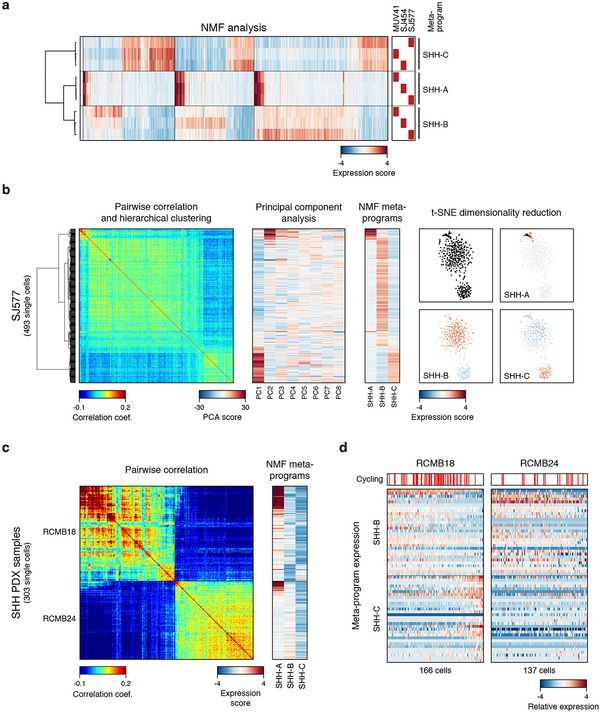
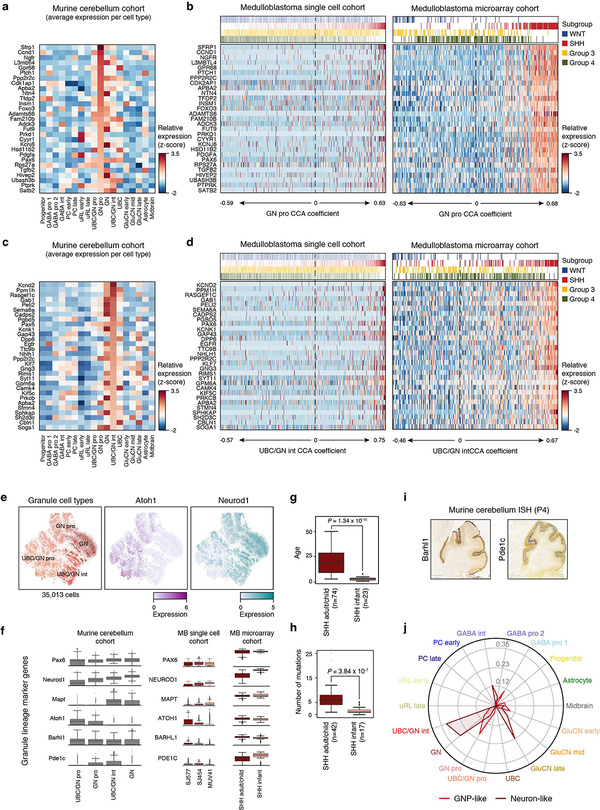
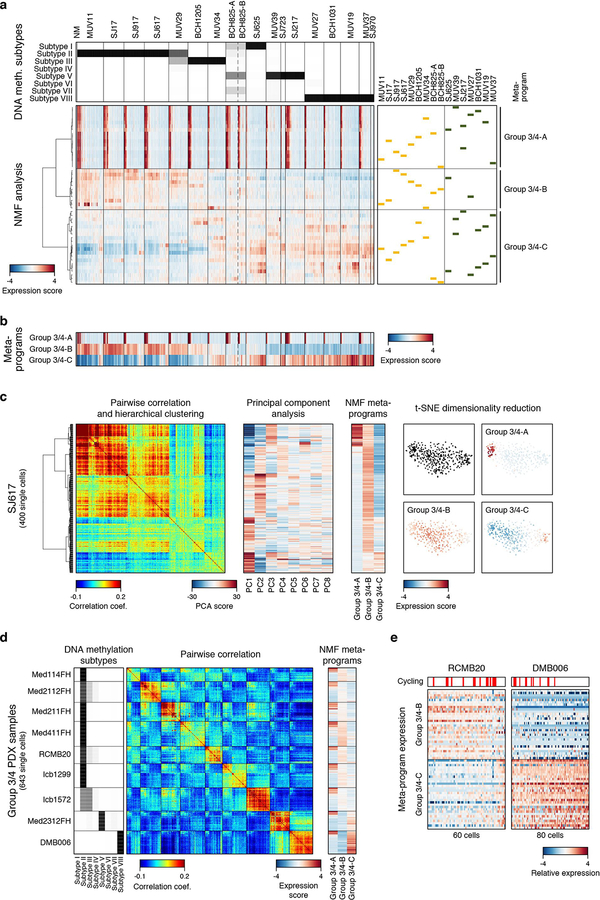
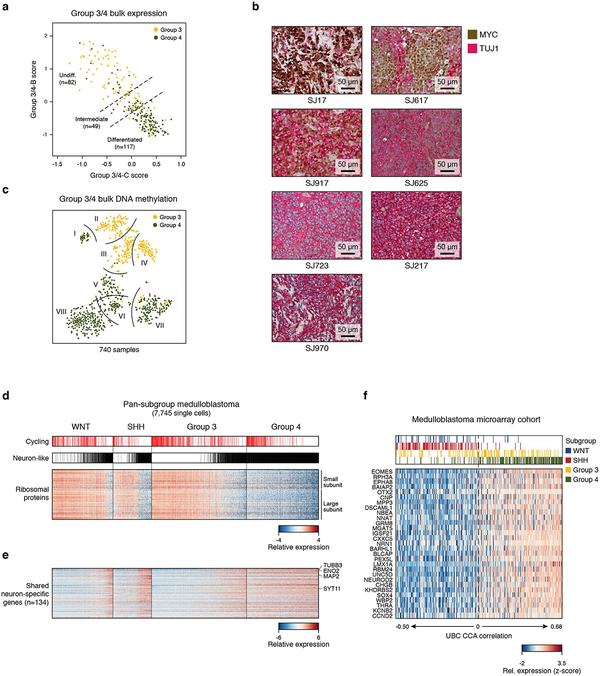
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
