

Impaired ligand-dependent MET activation caused by an extracellular SEMA domain missense mutation in lung cancer
- PMID: 31342590
- PMCID: PMC6778621
- DOI: 10.1111/cas.14142
Impaired ligand-dependent MET activation caused by an extracellular SEMA domain missense mutation in lung cancer
Abstract
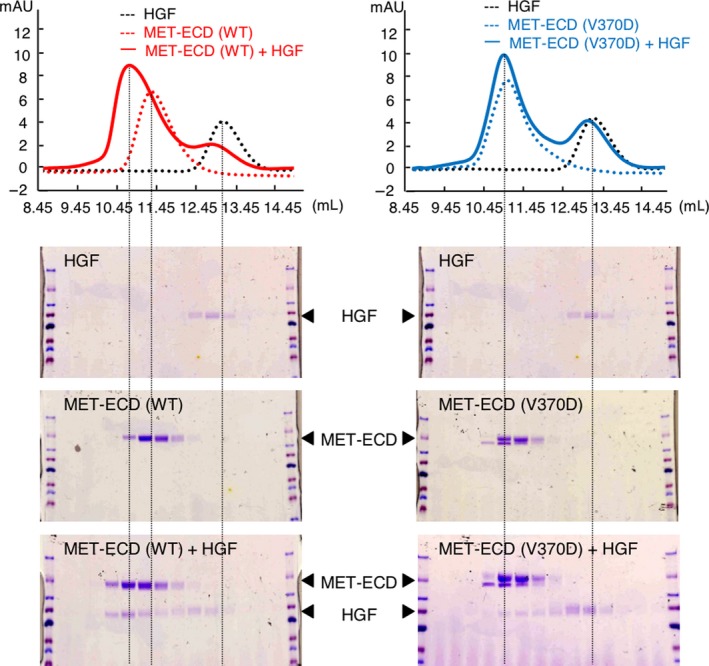
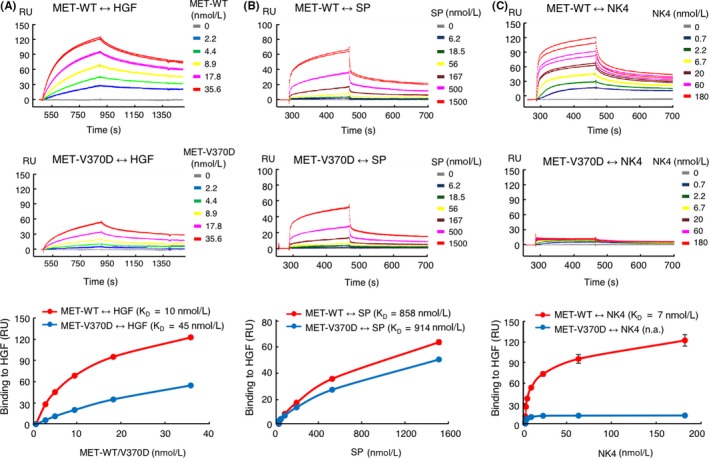
Aberrant activation of the MET/hepatocyte growth factor (HGF) receptor participates in the malignant behavior of cancer cells, such as invasion-metastasis and resistance to molecular targeted drugs. Many mutations in the MET extracellular region have been reported, but their significance is largely unknown. Here, we report the dysregulation of mutant MET originally found in a lung cancer patient with Val370 to Asp370 (V370D) replacement located in the extracellular SEMA domain. MET-knockout cells were prepared and reconstituted with WT-MET or V370D-MET. HGF stimulation induced MET dimerization and biological responses in cells reconstituted with WT-MET, but HGF did not induce MET dimerization and failed to induce biological responses in V370D-MET cells. The V370D mutation abrogated HGF-dependent drug resistance of lung cancer cells to epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKI). Compared with WT-MET cells, V370D-MET cells showed different activation patterns in receptor tyrosine kinases upon exposure to survival/growth-stressed conditions. Surface plasmon resonance analysis indicated that affinity between the extracellular region of V370D-MET and HGF was reduced compared with that for WT-MET. Further analysis of the association between V370D-MET and the separate domains of HGF indicated that the SP domain of HGF was unchanged, but its association with the NK4 domain of HGF was mostly lost in V370D-MET. These results indicate that the V370D mutation in the MET receptor impairs the functional association with HGF and is therefore a loss-of-function mutation. This mutation may change the dependence of cancer cell growth/survival on signaling molecules, which may promote cancer cell characteristics under certain conditions.
Keywords: MET receptor; affinity; hepatocyte growth factor; loss of function; missense mutation.
© 2019 The Authors. Cancer Science published by John Wiley & Sons Australia, Ltd on behalf of Japanese Cancer Association.
Conflict of interest statement
Authors declare no conflicts of interest for this article.
Figures
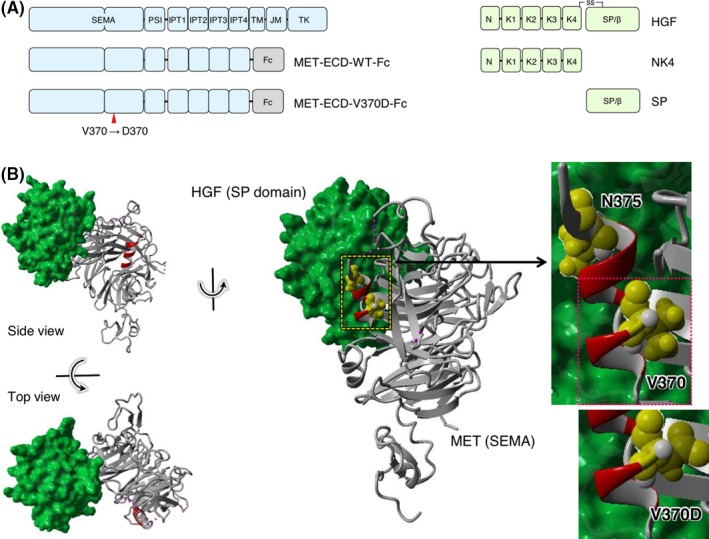
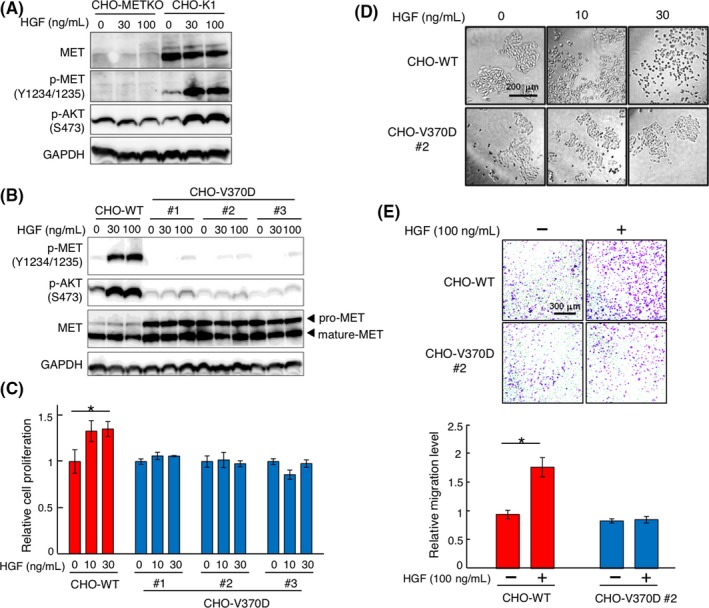
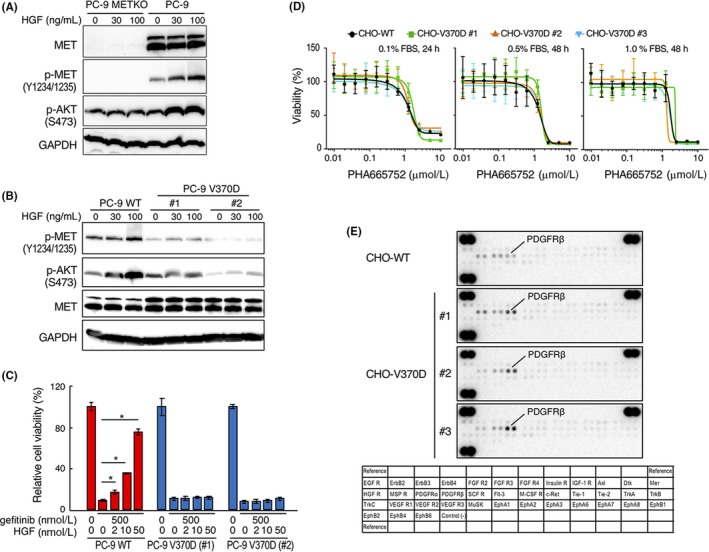
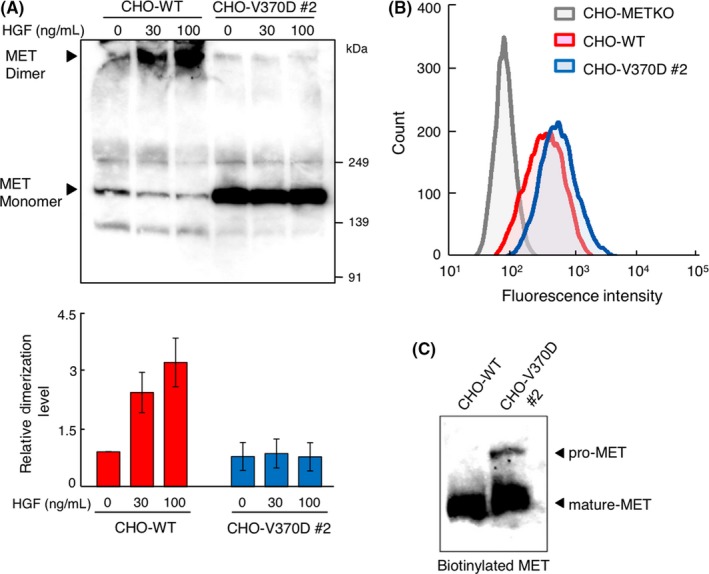
References
-
- Comoglio PM, Trusolino L, Boccaccio C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat Rev Cancer. 2018;18:341‐358. - PubMed
-
- Komada M, Hatsuzawa K, Shibamoto S, Ito F, Nakayama K, Kitamura N. Proteolytic processing of the hepatocyte growth factor/scatter factor receptor by furin. FEBS Lett. 1993;328:25‐29. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
