

Before and After: Comparison of Legacy and Harmonized TCGA Genomic Data Commons' Data
- PMID: 31344359
- PMCID: PMC6707074
- DOI: 10.1016/j.cels.2019.06.006
Before and After: Comparison of Legacy and Harmonized TCGA Genomic Data Commons' Data
Abstract
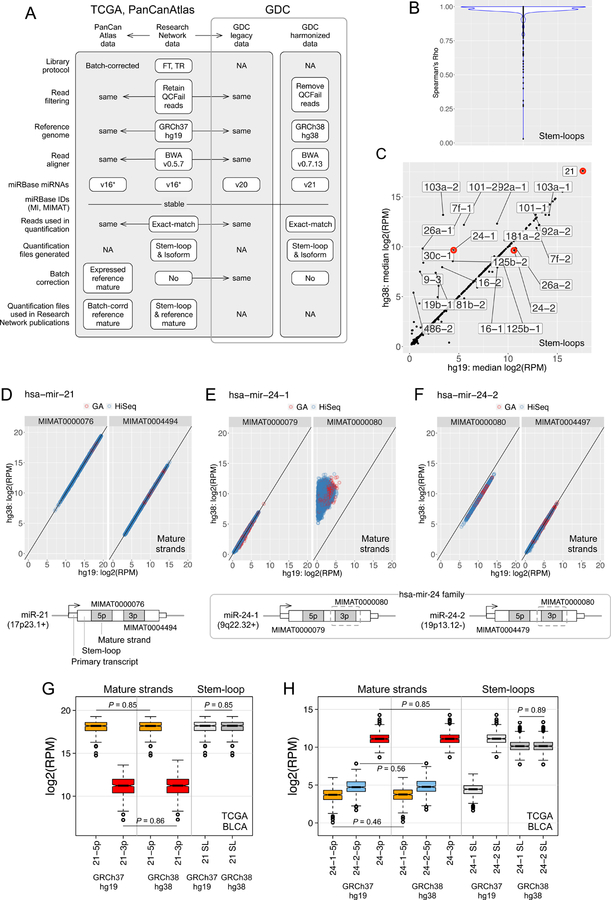
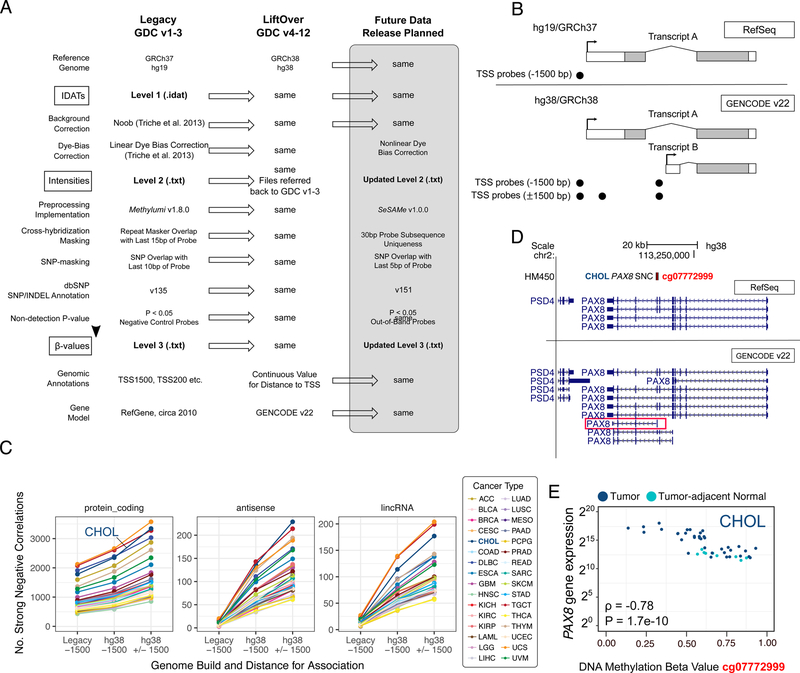
We present a systematic analysis of the effects of synchronizing a large-scale, deeply characterized, multi-omic dataset to the current human reference genome, using updated software, pipelines, and annotations. For each of 5 molecular data platforms in The Cancer Genome Atlas (TCGA)-mRNA and miRNA expression, single nucleotide variants, DNA methylation and copy number alterations-comprehensive sample, gene, and probe-level studies were performed, towards quantifying the degree of similarity between the 'legacy' GRCh37 (hg19) TCGA data and its GRCh38 (hg38) version as 'harmonized' by the Genomic Data Commons. We offer gene lists to elucidate differences that remained after controlling for confounders, and strategies to mitigate their impact on biological interpretation. Our results demonstrate that the hg19 and hg38 TCGA datasets are very highly concordant, promote informed use of either legacy or harmonized omics data, and provide a rubric that encourages similar comparisons as new data emerge and reference data evolve.
Keywords: DNA methylation; The Cancer Genome Atlas; human reference genome; mRNA expression; microRNA expression; quality control; somatic copy number alteration; somatic mutation.
Copyright © 2019 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests
The authors declare no competing interests.
Figures



References
-
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. (2011). High density DNA methylation array with single CpG site resolution. Genomics 98, 288–295. - PubMed
-
- Bowen NJ, Logani S, Dickerson EB, Kapa LB, Akhtar M, Benigno BB, and McDonald JF (2007). Emerging roles for PAX8 in ovarian cancer and endosalpingeal development. Gynecol Oncol 104, 331–337. - PubMed
-
- Brinkmann U, Vasmatzis G, Lee B, and Pastan I (1999). Novel genes in the PAGE and GAGE family of tumor antigens found by homology walking in the dbEST database. Cancer Res 59, 1445–1448. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U24 CA210978/CA/NCI NIH HHS/United States
- U24 CA210950/CA/NCI NIH HHS/United States
- U24 CA210974/CA/NCI NIH HHS/United States
- U24 CA210989/CA/NCI NIH HHS/United States
- U24 CA210952/CA/NCI NIH HHS/United States
- U24 CA210957/CA/NCI NIH HHS/United States
- R01 CA175486/CA/NCI NIH HHS/United States
- U24 CA210949/CA/NCI NIH HHS/United States
- U24 CA209851/CA/NCI NIH HHS/United States
- U24 CA210990/CA/NCI NIH HHS/United States
- U24 CA211000/CA/NCI NIH HHS/United States
- P30 ES010126/ES/NIEHS NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- U24 CA210969/CA/NCI NIH HHS/United States
- U24 CA210988/CA/NCI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- U24 CA211006/CA/NCI NIH HHS/United States
- U24 CA210999/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
