

Acquired temozolomide resistance in MGMT-deficient glioblastoma cells is associated with regulation of DNA repair by DHC2
- PMID: 31347685
- PMCID: PMC6658867
- DOI: 10.1093/brain/awz202
Acquired temozolomide resistance in MGMT-deficient glioblastoma cells is associated with regulation of DNA repair by DHC2
Abstract
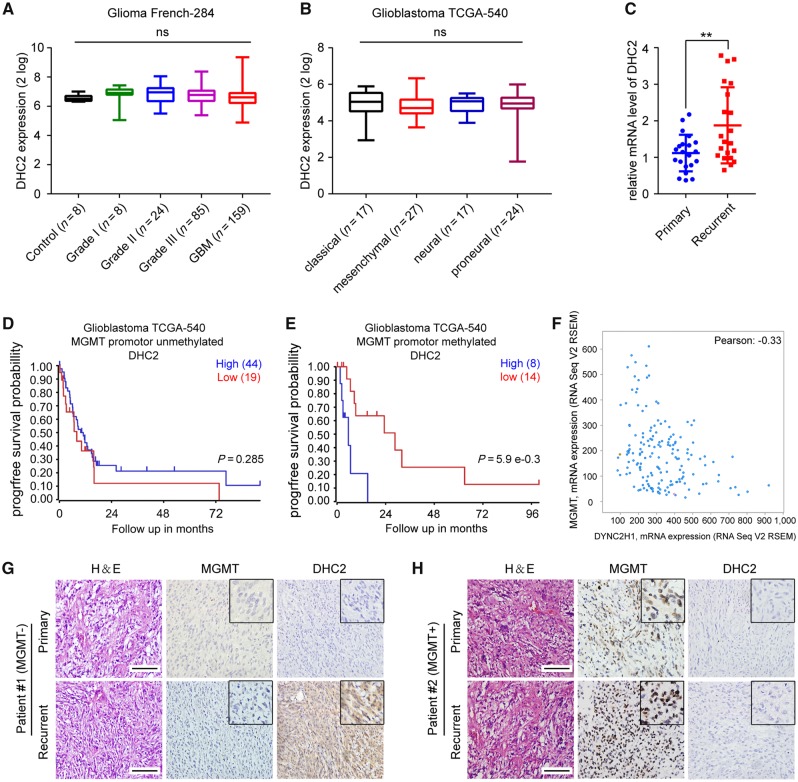
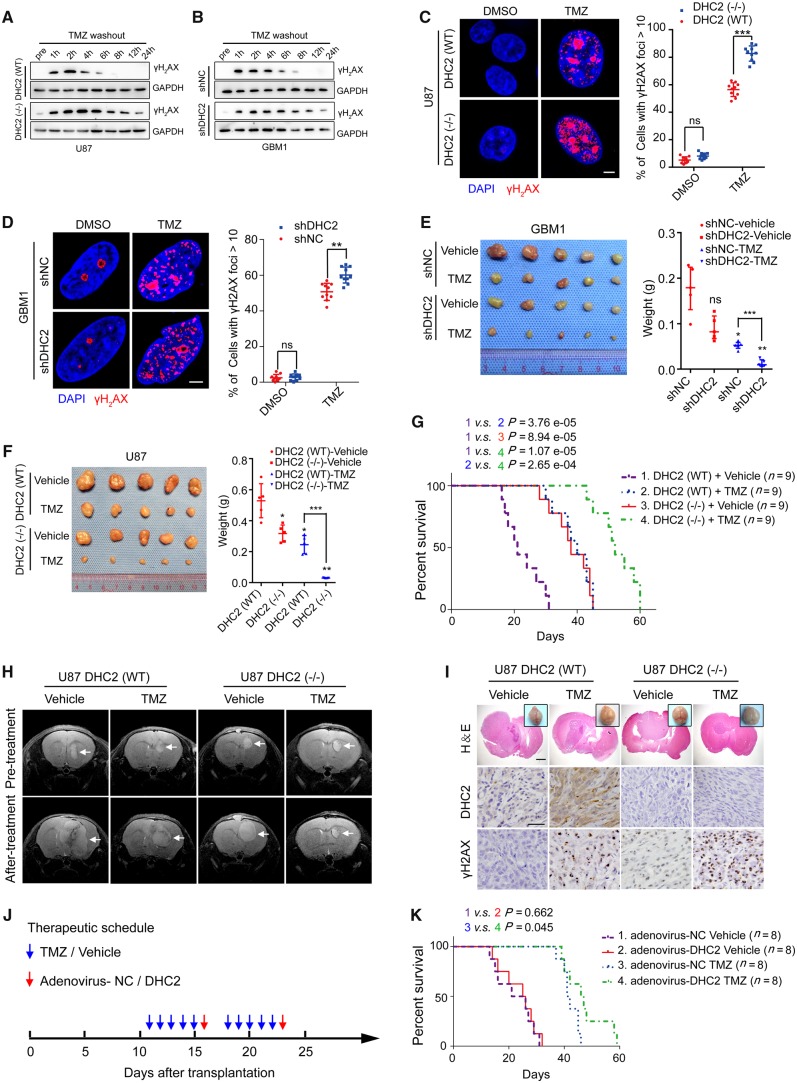
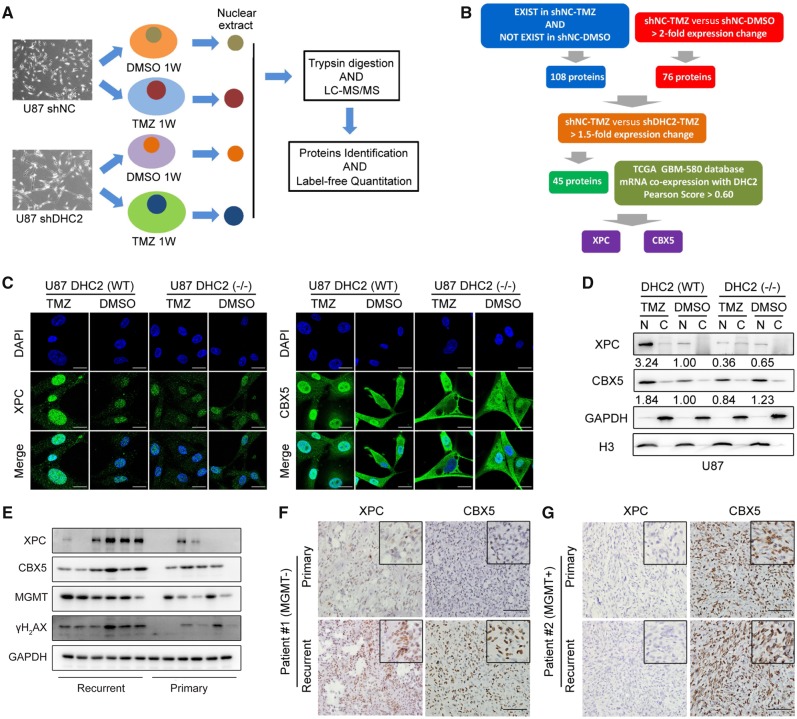
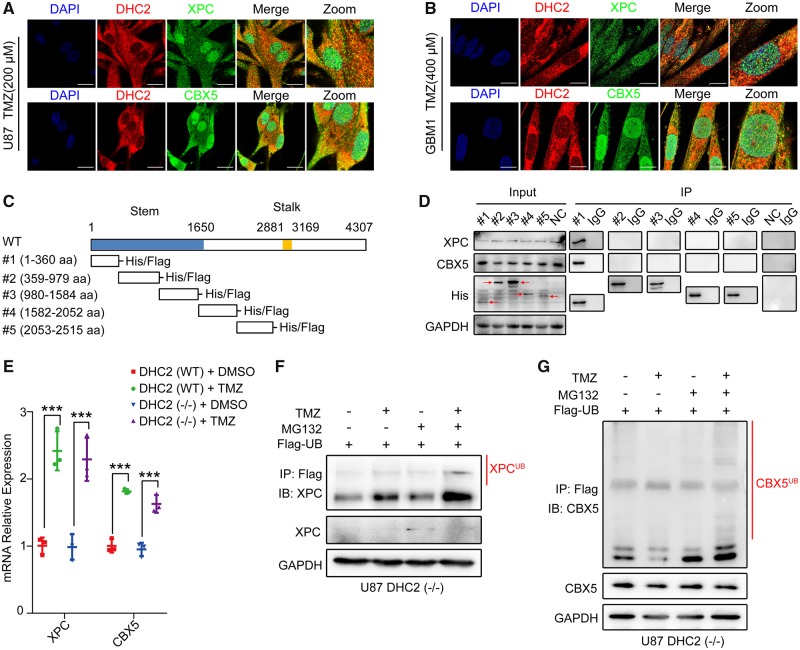
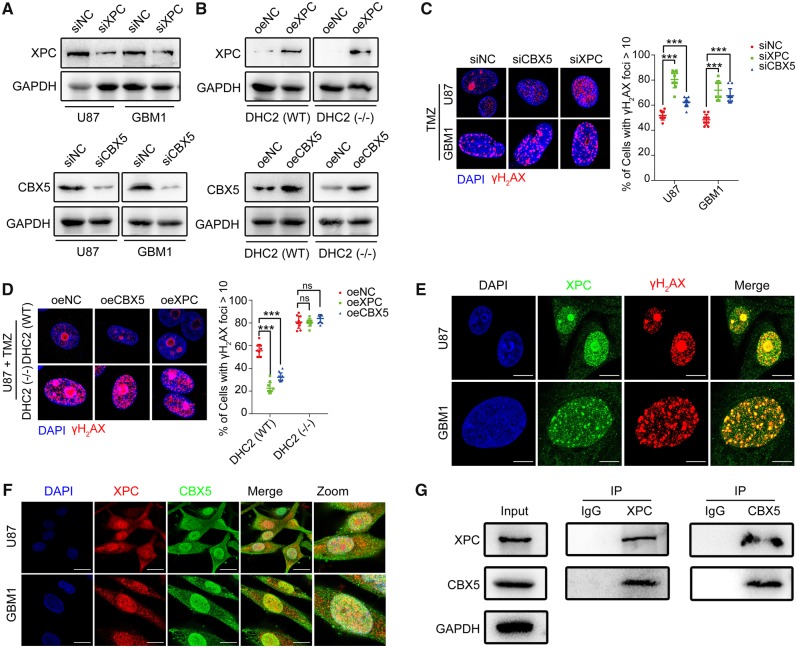
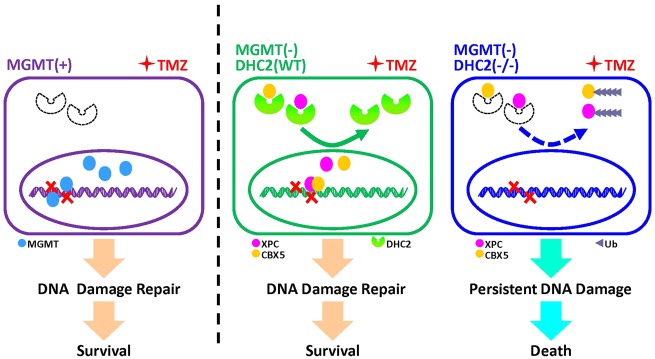
The acquisition of temozolomide resistance is a major clinical challenge for glioblastoma treatment. Chemoresistance in glioblastoma is largely attributed to repair of temozolomide-induced DNA lesions by O6-methylguanine-DNA methyltransferase (MGMT). However, some MGMT-deficient glioblastomas are still resistant to temozolomide, and the underlying molecular mechanisms remain unclear. We found that DYNC2H1 (DHC2) was expressed more in MGMT-deficient recurrent glioblastoma specimens and its expression strongly correlated to poor progression-free survival in MGMT promotor methylated glioblastoma patients. Furthermore, silencing DHC2, both in vitro and in vivo, enhanced temozolomide-induced DNA damage and significantly improved the efficiency of temozolomide treatment in MGMT-deficient glioblastoma. Using a combination of subcellular proteomics and in vitro analyses, we showed that DHC2 was involved in nuclear localization of the DNA repair proteins, namely XPC and CBX5, and knockdown of either XPC or CBX5 resulted in increased temozolomide-induced DNA damage. In summary, we identified the nuclear transportation of DNA repair proteins by DHC2 as a critical regulator of acquired temozolomide resistance in MGMT-deficient glioblastoma. Our study offers novel insights for improving therapeutic management of MGMT-deficient glioblastoma.
Keywords: CBX5; DHC2; XPC; acquired TMZ resistance; glioblastoma.
© The Author(s) (2019). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
References
-
- Batista LF, Roos WP, Christmann M, Menck CF, Kaina B. Differential sensitivity of malignant glioma cells to methylating and chloroethylating anticancer drugs: p53 determines the switch by regulating xpc, ddb2, and DNA double-strand breaks. Cancer Res 2007; 67: 11886–95. - PubMed
-
- Batty D, Rapic’-Otrin V, Levine AS, Wood RD. Stable binding of human XPC complex to irradiated DNA confers strong discrimination for damaged sites. J Mol Biol 2000; 300: 275–90. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
