

The presence and impact of reference bias on population genomic studies of prehistoric human populations
- PMID: 31348818
- PMCID: PMC6685638
- DOI: 10.1371/journal.pgen.1008302
The presence and impact of reference bias on population genomic studies of prehistoric human populations
Abstract
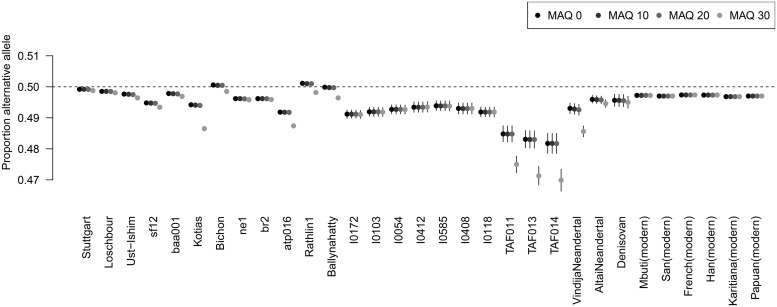
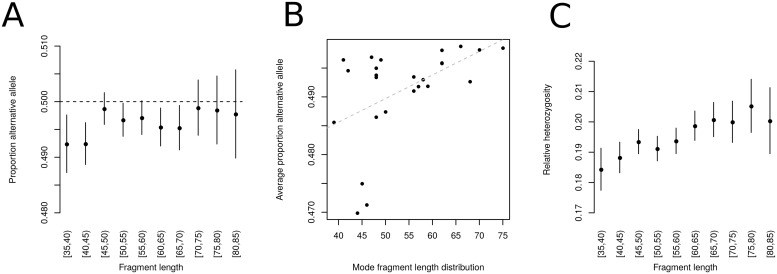
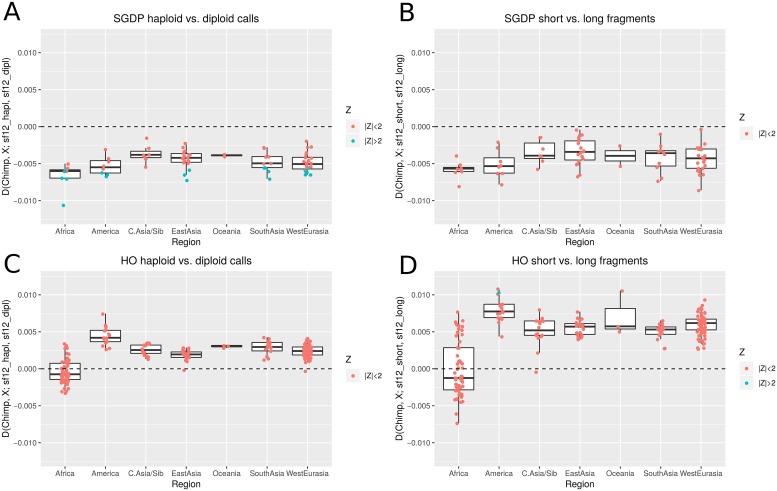
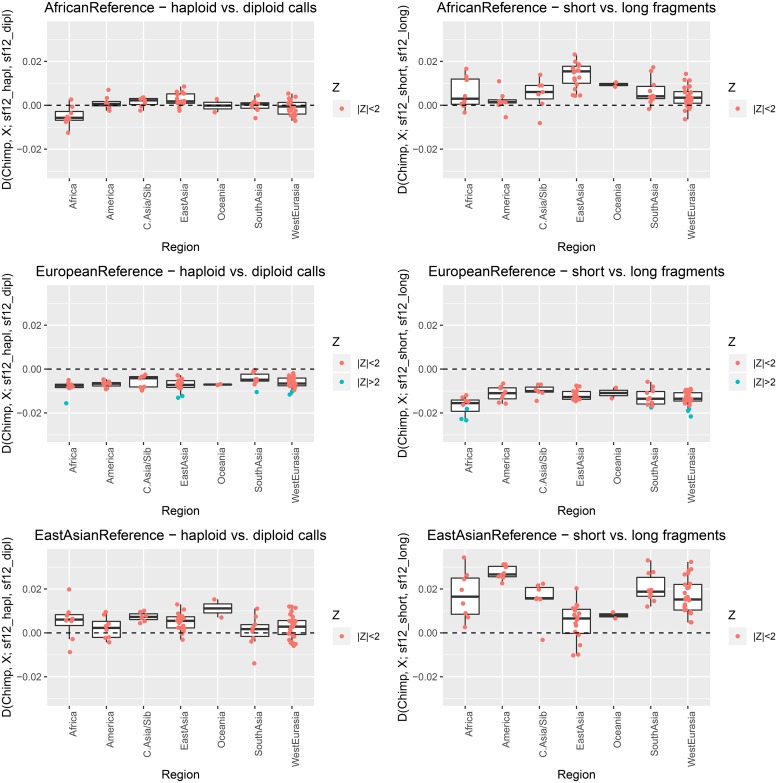
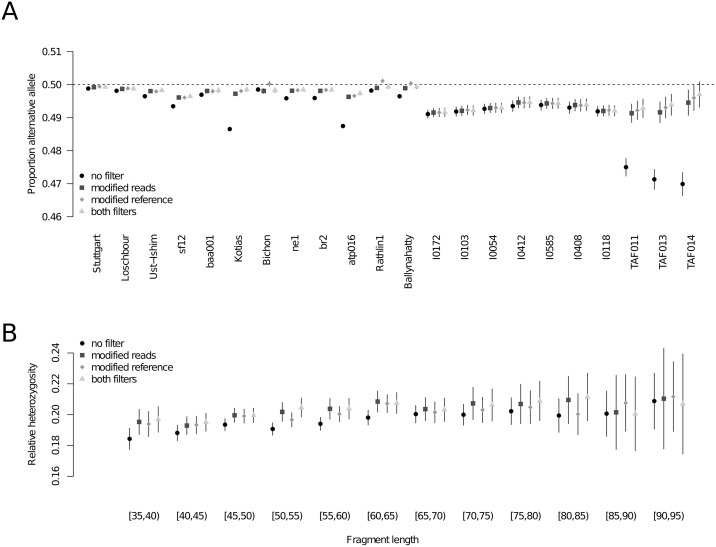
Haploid high quality reference genomes are an important resource in genomic research projects. A consequence is that DNA fragments carrying the reference allele will be more likely to map successfully, or receive higher quality scores. This reference bias can have effects on downstream population genomic analysis when heterozygous sites are falsely considered homozygous for the reference allele. In palaeogenomic studies of human populations, mapping against the human reference genome is used to identify endogenous human sequences. Ancient DNA studies usually operate with low sequencing coverages and fragmentation of DNA molecules causes a large proportion of the sequenced fragments to be shorter than 50 bp-reducing the amount of accepted mismatches, and increasing the probability of multiple matching sites in the genome. These ancient DNA specific properties are potentially exacerbating the impact of reference bias on downstream analyses, especially since most studies of ancient human populations use pseudo-haploid data, i.e. they randomly sample only one sequencing read per site. We show that reference bias is pervasive in published ancient DNA sequence data of prehistoric humans with some differences between individual genomic regions. We illustrate that the strength of reference bias is negatively correlated with fragment length. Most genomic regions we investigated show little to no mapping bias but even a small proportion of sites with bias can impact analyses of those particular loci or slightly skew genome-wide estimates. Therefore, reference bias has the potential to cause minor but significant differences in the results of downstream analyses such as population allele sharing, heterozygosity estimates and estimates of archaic ancestry. These spurious results highlight how important it is to be aware of these technical artifacts and that we need strategies to mitigate the effect. Therefore, we suggest some post-mapping filtering strategies to resolve reference bias which help to reduce its impact substantially.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Bobo D, Lipatov M, Rodriguez-Flores JL, Auton A, Henn BM. False Negatives Are a Significant Feature of Next Generation Sequencing Callsets. bioRxiv. 2016; p. 066043.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
