

Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis
- PMID: 31348891
- PMCID: PMC6662628
- DOI: 10.1016/j.cell.2019.06.029
Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis
Abstract
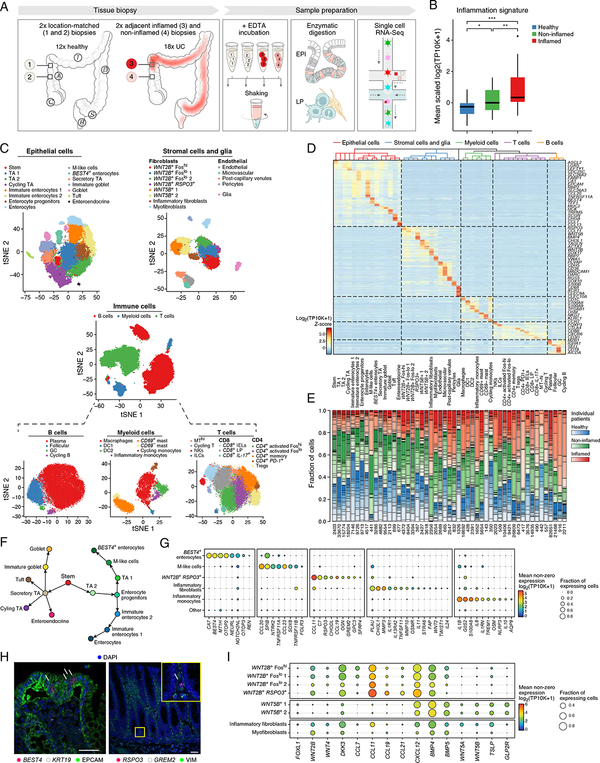
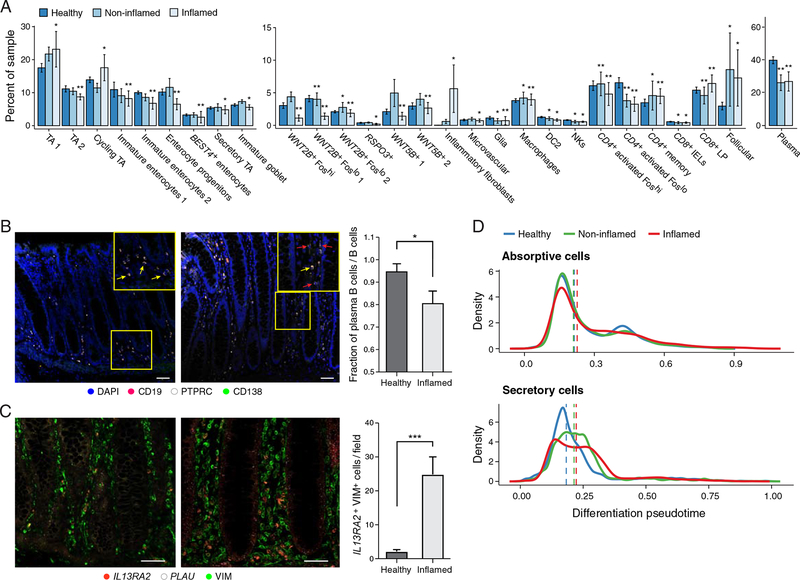
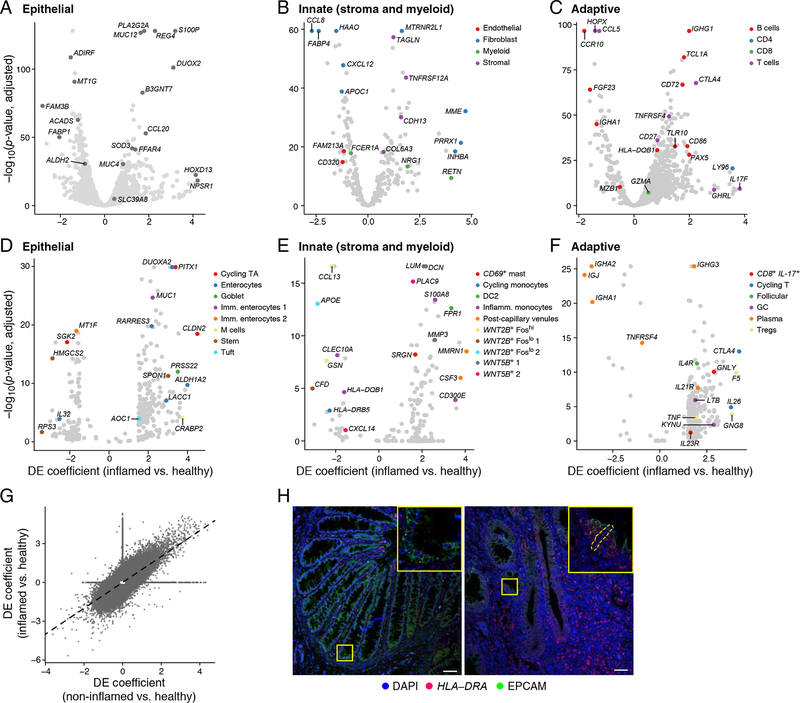
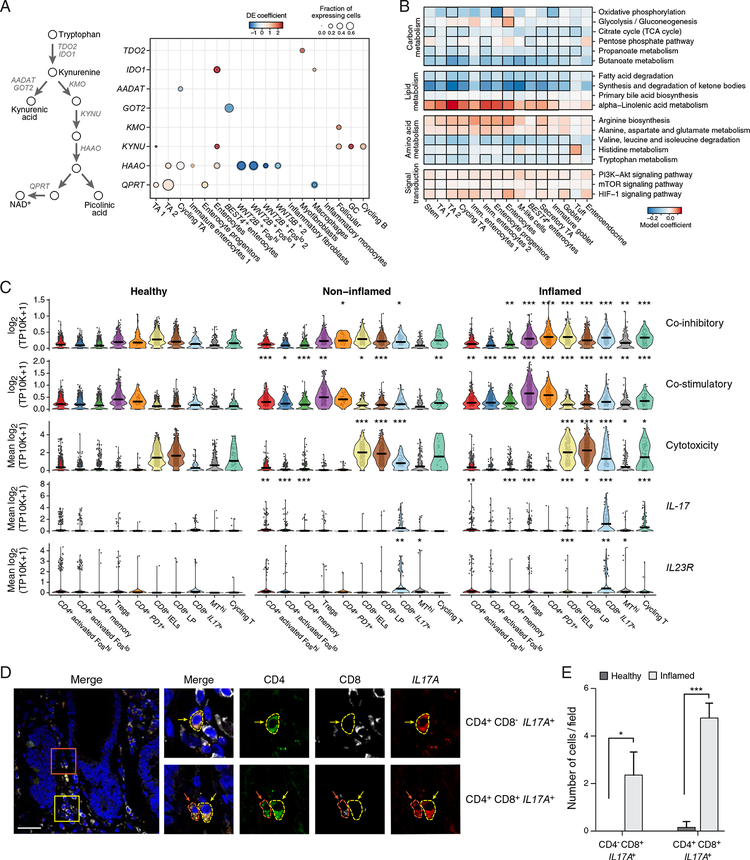
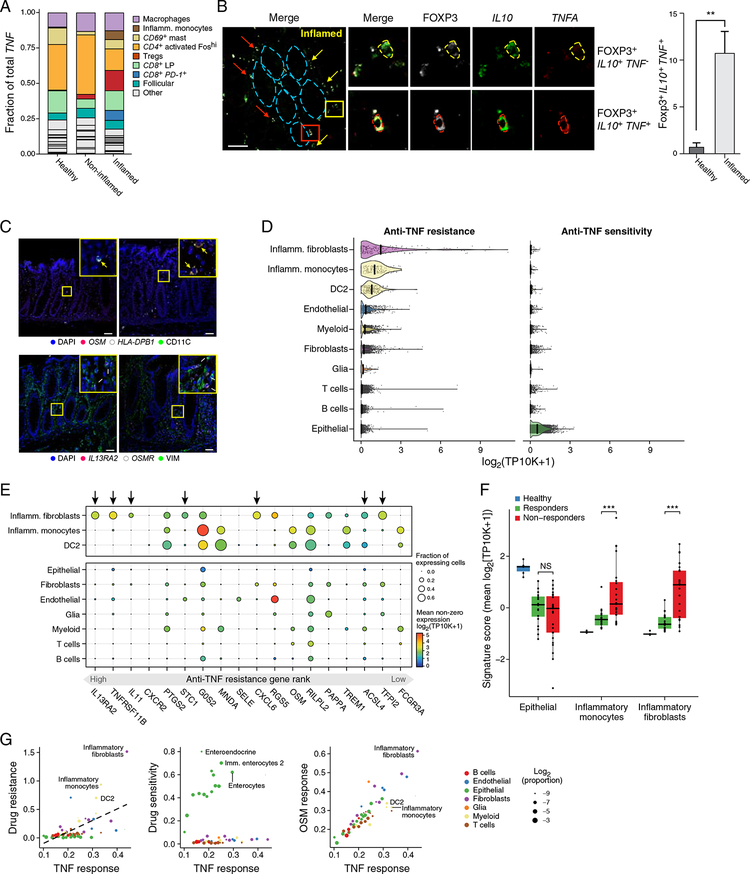
Genome-wide association studies (GWAS) have revealed risk alleles for ulcerative colitis (UC). To understand their cell type specificities and pathways of action, we generate an atlas of 366,650 cells from the colon mucosa of 18 UC patients and 12 healthy individuals, revealing 51 epithelial, stromal, and immune cell subsets, including BEST4+ enterocytes, microfold-like cells, and IL13RA2+IL11+ inflammatory fibroblasts, which we associate with resistance to anti-TNF treatment. Inflammatory fibroblasts, inflammatory monocytes, microfold-like cells, and T cells that co-express CD8 and IL-17 expand with disease, forming intercellular interaction hubs. Many UC risk genes are cell type specific and co-regulated within relatively few gene modules, suggesting convergence onto limited sets of cell types and pathways. Using this observation, we nominate and infer functions for specific risk genes across GWAS loci. Our work provides a framework for interrogating complex human diseases and mapping risk variants to cell types and pathways.
Keywords: anti-TNF resistance; cell-cell interactions; colon; genome-wide association studies; inflammation; inflammatory bowel disease; large intestine; single-cell RNA-seq; single-cell genomics; ulcerative colitis.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
A.R. is an SAB member of ThermoFisher Scientific and Syros Pharmaceuticals. A.R. and R.J.X. are cofounders of and equity holders in Celsius Therapeutics. R.J.X. is a consultant to Novartis. A.K.S was compensated for consulting and SAB membership from Honeycomb Biotechnologies, Dot Bio, Cellarity, Cogen Therapeutics, and Dahlia Biosciences. M.B., A.L.H., N.R., R.H.H., J.O.-M., O.R.-R., A.K.S., K.S., C.S.S., A.R. and R.J.X. are co-inventors on PCT/US2018/042554 relating to advances in understanding cellular dynamics, cellular effectors, and risk variants of the human colon in health and UC described in this manuscript.
Processed data was deposited in the Single Cell Portal (SCP259). Raw data will be available for download from Broad DUOS.
Figures


Comment in
-
The Cell Circuitry of Ulcerative Colitis, a New View for a Highly Complex Disease.Gastroenterology. 2020 Apr;158(5):1506-1508. doi: 10.1053/j.gastro.2020.02.019. Epub 2020 Feb 15. Gastroenterology. 2020. PMID: 32068021 Free PMC article. No abstract available.
References
-
- Arijs I, Li K, Toedter G, Quintens R, Van Lommel L, Van Steen K, Leemans P, De Hertogh G, Lemaire K, Ferrante M, et al. (2009). Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut 58, 1612–1619. - PubMed
-
- Atreya R, Zimmer M, Bartsch B, Waldner MJ, Atreya I, Neumann H, Hildner K, Hoffman A, Kiesslich R, Rink AD, et al. (2011). Antibodies against tumor necrosis factor (TNF) induce T-cell apoptosis in patients with inflammatory bowel diseases via TNF receptor 2 and intestinal CD14(+) macrophages. Gastroenterology 141, 2026–2038. - PubMed
-
- Bastian M, Heymann S, and Jacomy M (2009). Gephi: An Open Source Software for Exploring and Manipulating Networks.
-
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 57, 289–300.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
