

Somatic mosaicism of sex chromosomes in the blood and brain
- PMID: 31348909
- PMCID: PMC6717667
- DOI: 10.1016/j.brainres.2019.146345
Somatic mosaicism of sex chromosomes in the blood and brain
Abstract
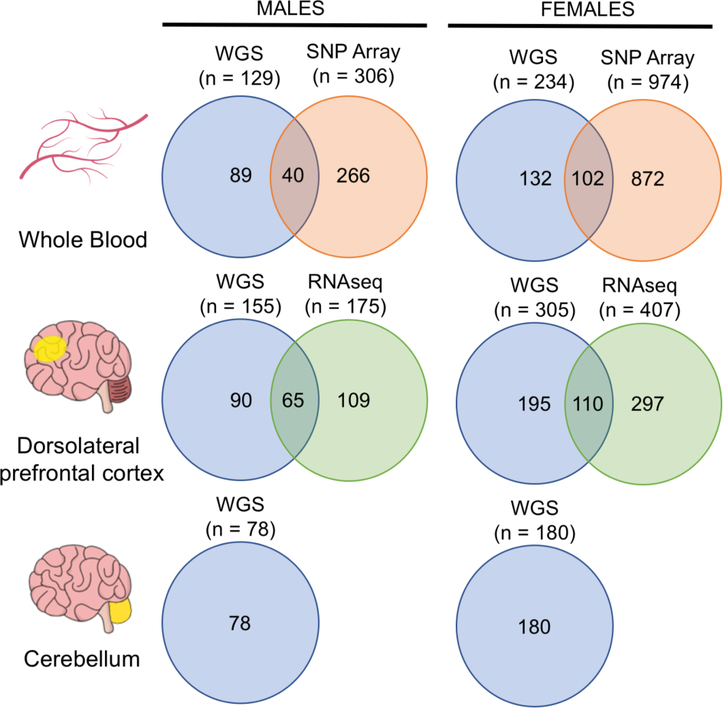
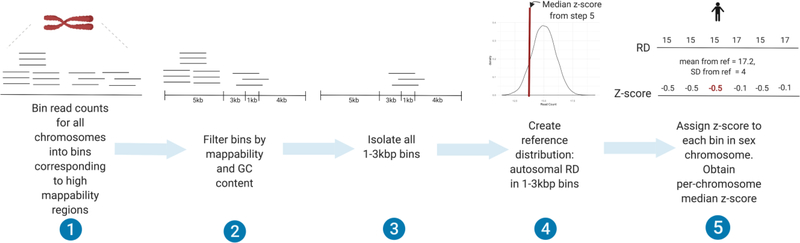
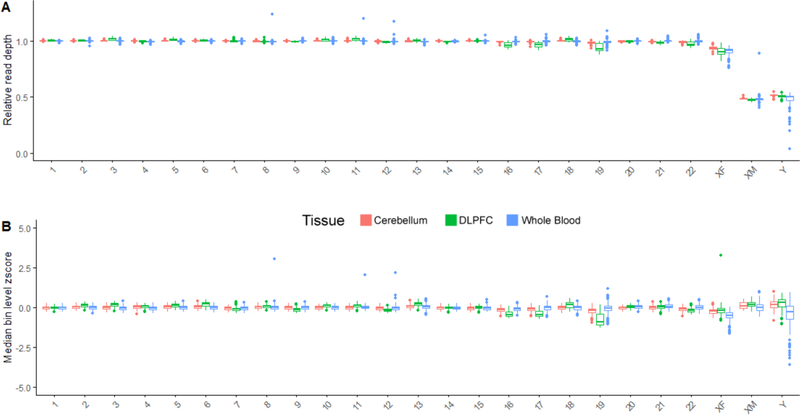
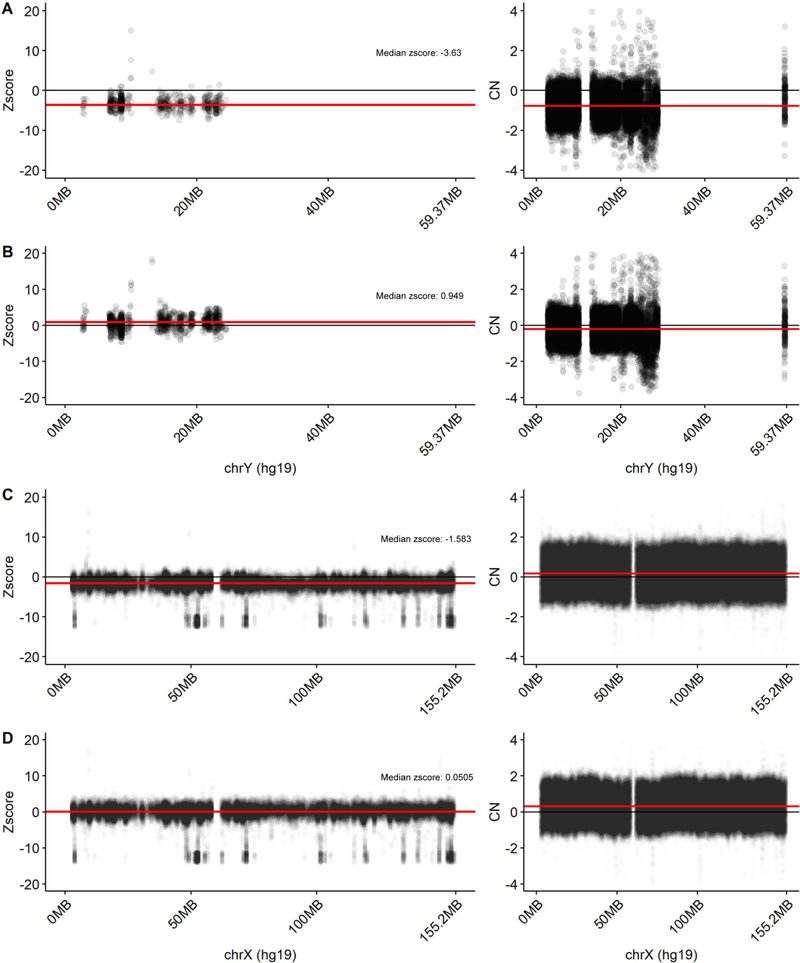
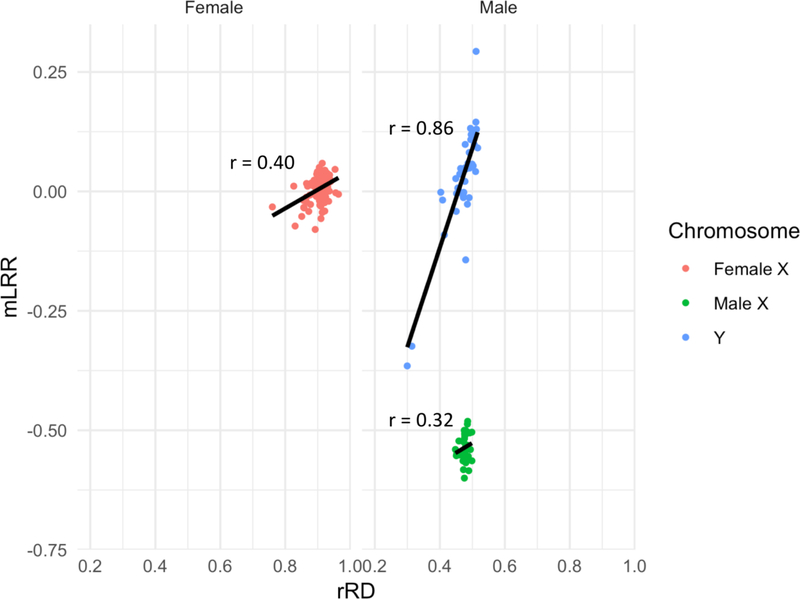
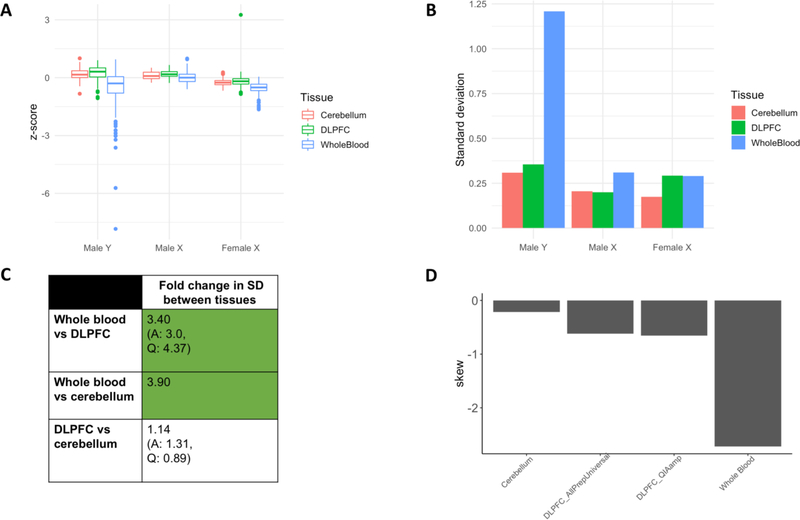
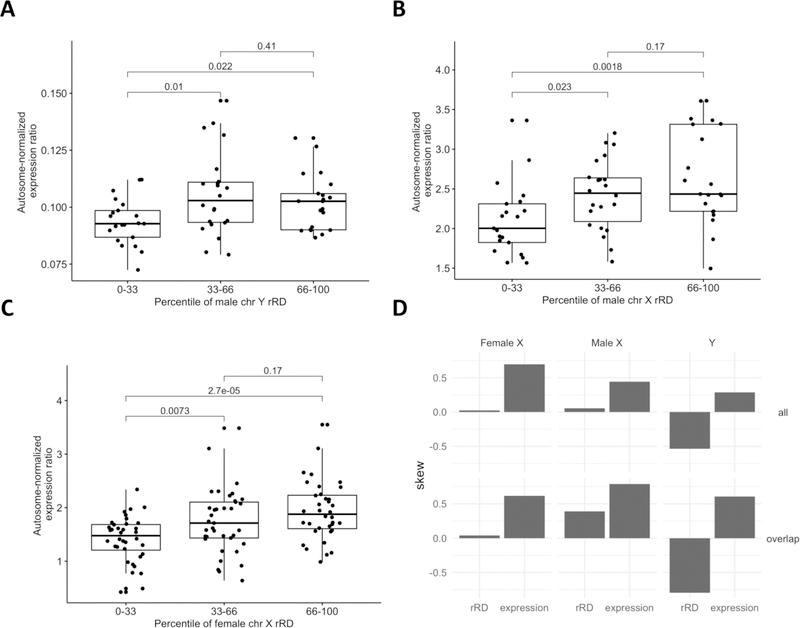
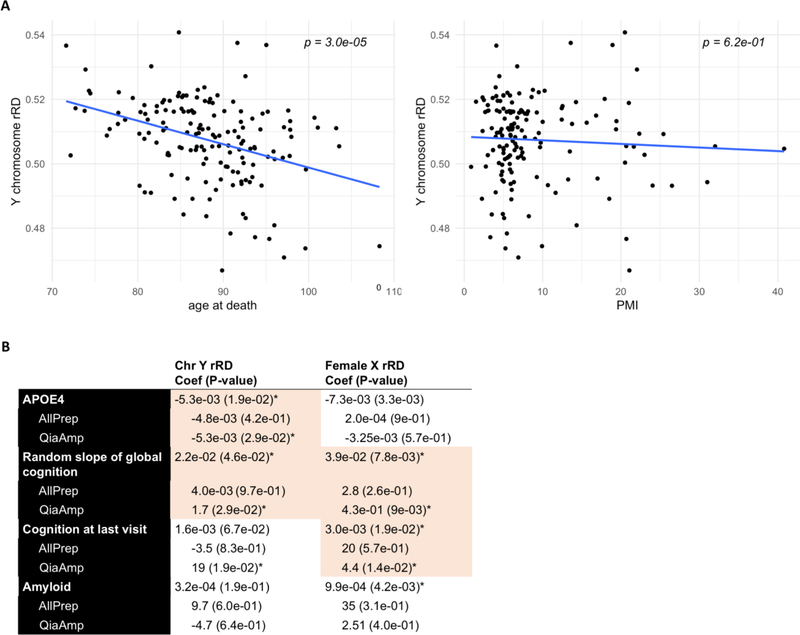
In the blood, mosaic somatic aneuploidy (mSA) of all chromosomes has been found to be associated with adverse health outcomes, including hematological cancer. Sex chromosome mSA in the blood has been found to occur at a higher rate than autosomal mSA. Mosaic loss of the Y chromosome is the most common copy number alteration in males, and has been found to be associated with Alzheimer's disease (AD) in blood lymphocytes. mSA of the sex chromosomes has also been identified in the brain; however, little is known about its frequency across individuals. Using WGS data from 362 males and 719 females from the ROSMAP cohort, we quantified the relative rate of sex chromosome mSA in the dorsolateral prefrontal cortex (DLPFC), cerebellum and whole blood. To ascertain the functionality of observed sex chromosome mosaicism in the DLPFC, we examined its correlation with chromosome X and Y gene expression as well as neuropathological and clinical characteristics of AD and cognitive ageing. In males, we found that mSA of the Y chromosome occurs more frequently in blood than in the DLPFC or cerebellum. In the DLPFC, the presence of at least one APOE4 allele was associated with a reduction in read depth of the Y chromosome (p = 1.9e-02). In the female DLPFC, a reduction in chromosome X read depth was associated with reduced cognition at the last clinical visit and faster rate of cognitive decline (p = 7.8e-03; p = 1.9e-02). mSA of all sex chromosomes in the DLPFC were associated with aggregate measures of gene expression, implying functional impact. Our results provide insight into the relative rate of mSA between tissues and suggest that Y and female X chromosome read depth in the DLPFC is modestly associated with late AD risk factors and cognitive pathologies.
Keywords: Alzheimer's disease; Aneuploidy; Sex differences; Somatic mosaicism; Whole genome sequencing.
Copyright © 2019 Elsevier B.V. All rights reserved.
Figures
References
-
- Arendt T, Bruckner MK, Losche A, 2015. Regional mosaic genomic heterogeneity in the elderly and in Alzheimer’s disease as a correlate of neuronal vulnerability. Acta Neuropathol. 130 pp. 501–510 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
