

Update on mutations in the HIF: EPO pathway and their role in erythrocytosis
- PMID: 31350093
- PMCID: PMC7484688
- DOI: 10.1016/j.blre.2019.100590
Update on mutations in the HIF: EPO pathway and their role in erythrocytosis
Abstract
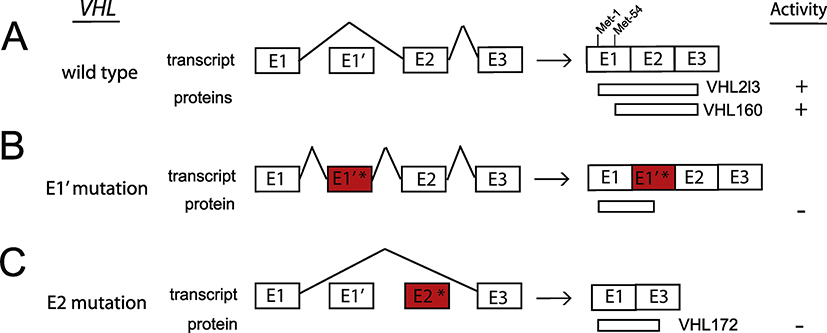
Identification of the underlying defects in congenital erythrocytosis has provided mechanistic insights into the regulation of erythropoiesis and oxygen homeostasis. The Hypoxia Inducible Factor (HIF) pathway plays a key role in this regard. In this pathway, an enzyme, Prolyl Hydroxylase Domain protein 2 (PHD2), constitutively prolyl hydroxylates HIF-2α, thereby targeting HIF-2α for degradation by the von Hippel Lindau (VHL) tumor suppressor protein. Under hypoxia, this modification is attenuated, resulting in the stabilization of HIF-2α and transcriptional activation of the erythropoietin (EPO) gene. Circulating EPO then binds to the EPO receptor (EPOR) on red cell progenitors in the bone marrow, leading to expansion of red cell mass. Loss of function mutations in PHD2 and VHL, as well as gain of function mutations in HIF-2α and EPOR, are well established causes of erythrocytosis. Here, we highlight recent developments that show that the study of this condition is still evolving. Specifically, novel mutations have been identified that either change amino acids in the zinc finger domain of PHD2 or alter splicing of the VHL gene. In addition, continued study of HIF-2α mutations has revealed a distinctive genotype-phenotype correlation. Finally, novel mutations have recently been identified in the EPO gene itself. Thus, the cascade of genes that at a molecular level leads to EPO action, namely PHD2 - > HIF2A - > VHL - > EPO - > EPOR, are all mutational targets in congenital erythrocytosis.
Keywords: Erythrocytosis; Erythropoiesis; Erythropoietin; Hypoxia Inducible Factor; Oxygen sensing; Polycythemia; Prolyl Hydroxylase Domain protein; Zinc finger; von Hippel Lindau tumor suppressor protein.
Copyright © 2019. Published by Elsevier Ltd.
Conflict of interest statement
Conflict of interest
No conflicts to disclose.
Figures





References
-
- Wenger RH, Hoogewijs D. Regulated oxygen sensing by protein hydroxylation in renal erythropoietin-producing cells. Am J Physiol Renal Physiol. 2010;298:F1287–96. - PubMed
-
- Suzuki N, Yamamoto M. Roles of renal erythropoietin-producing (REP) cells in the maintenance of systemic oxygen homeostasis. Pflugers Arch. 2016;468:3–12. - PubMed
-
- Kaelin WG Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
