

A machine-compiled database of genome-wide association studies
- PMID: 31350405
- PMCID: PMC6659642
- DOI: 10.1038/s41467-019-11026-x
A machine-compiled database of genome-wide association studies
Abstract
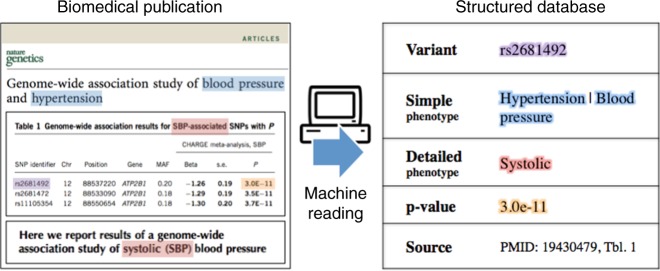
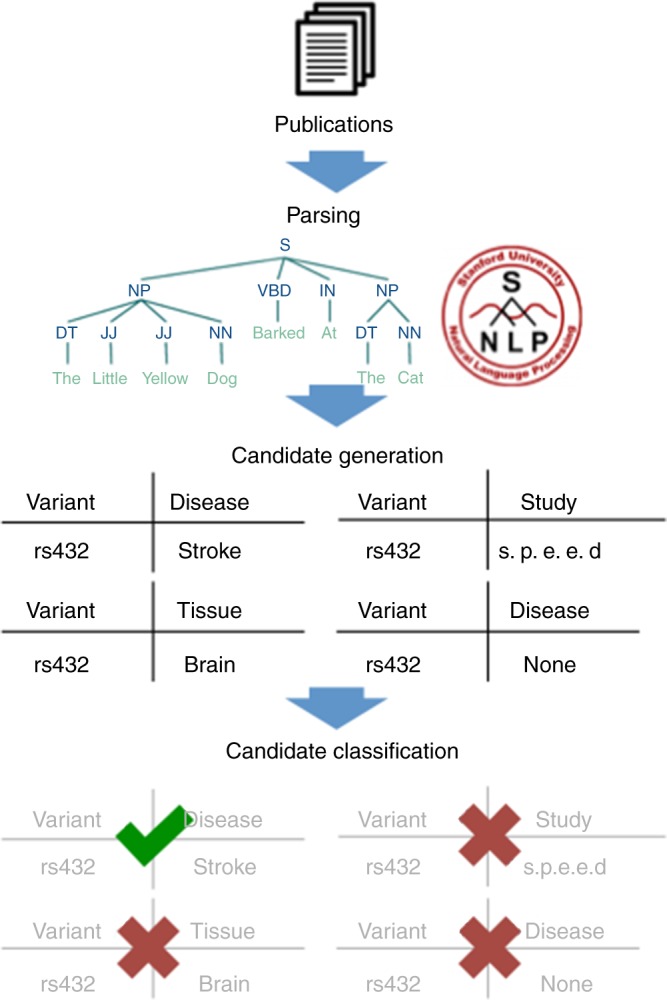
Tens of thousands of genotype-phenotype associations have been discovered to date, yet not all of them are easily accessible to scientists. Here, we describe GWASkb, a machine-compiled knowledge base of genetic associations collected from the scientific literature using automated information extraction algorithms. Our information extraction system helps curators by automatically collecting over 6,000 associations from open-access publications with an estimated recall of 60-80% and with an estimated precision of 78-94% (measured relative to existing manually curated knowledge bases). This system represents a fully automated GWAS curation effort and is made possible by a paradigm for constructing machine learning systems called data programming. Our work represents a step towards making the curation of scientific literature more efficient using automated systems.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Promethease. https://promethease.com/ (2019)
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
