

Genotype-phenotype associations in Fanconi anemia: A literature review
- PMID: 31351673
- PMCID: PMC6730648
- DOI: 10.1016/j.blre.2019.100589
Genotype-phenotype associations in Fanconi anemia: A literature review
Abstract
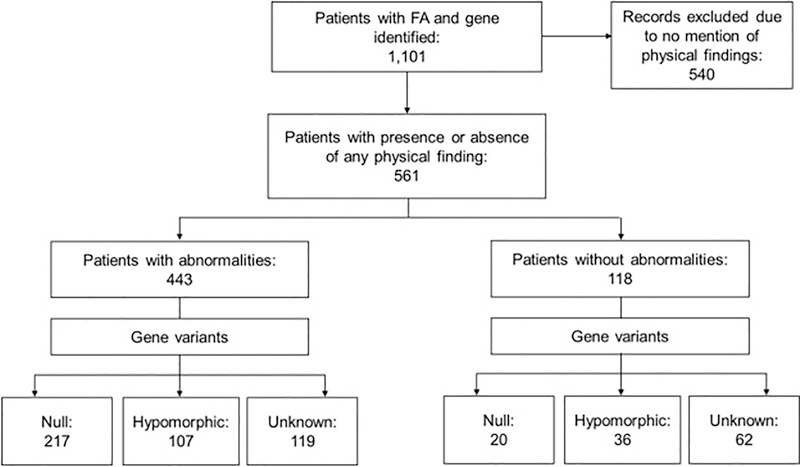
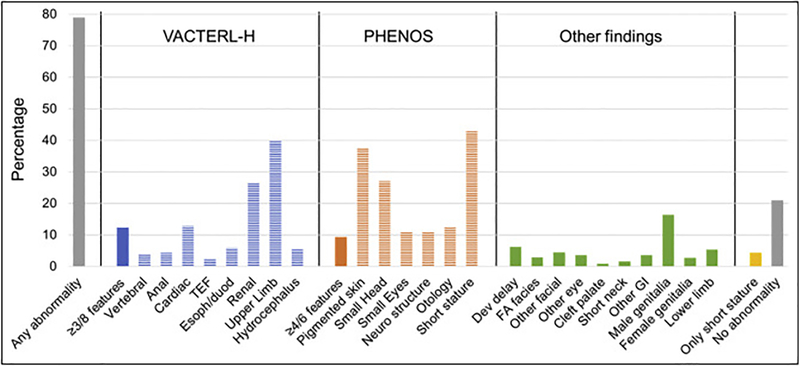
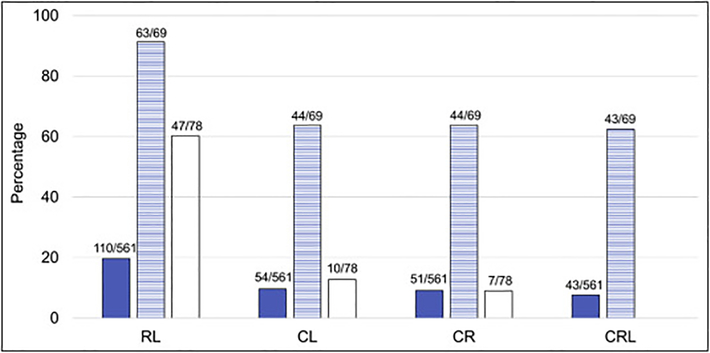
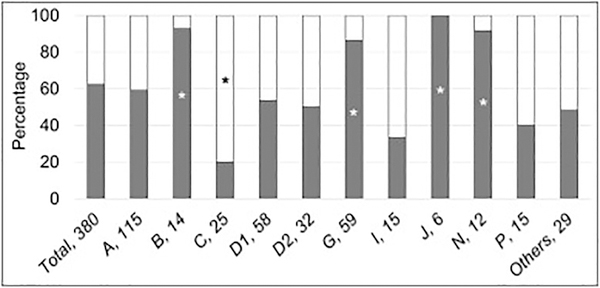
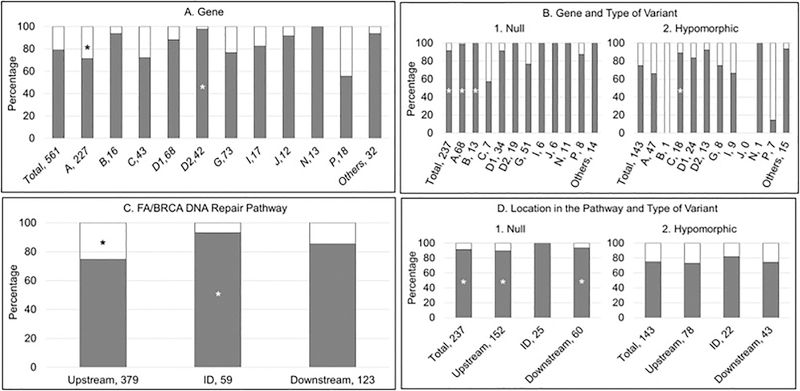
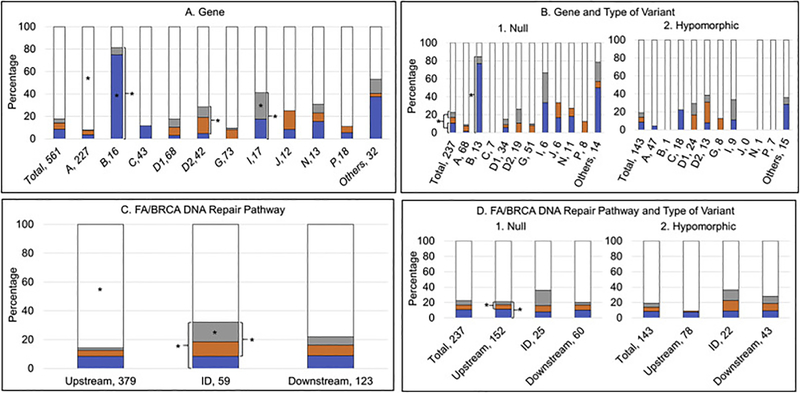
Fanconi anemia (FA) is a genomic instability syndrome with predisposition to congenital abnormalities, bone marrow failure, and cancer. Classical and most frequent congenital abnormalities include all those seen in VACTERL-H association and those described under the PHENOS acronym. Pathogenic variants in at least 22 genes are associated with FA, which code for proteins that comprise the FA/BRCA DNA repair pathway. We reviewed 187 publications and 1101 cases of FA in which the gene or complementation group was identified and analyzed those in whom physical findings were sought. We conducted genotype-phenotype analyses considering the specific gene, the location in the FA/BRCA DNA repair pathway, and the type of variant (null or hypomorphic) as exposures. The outcomes were the presence of any physical abnormality or specific categories of abnormalities. Seventy-nine percent of the patients had at least one physical abnormality. Pathogenic variants in FANCB, FANCD2, the ID complex and downstream genes were associated with several specific anomalies. Patients with biallelic or hemizygous null variants had a higher proportion of at least one abnormality, renal malformations, microcephaly, short stature and the combination of VACTERL-H compared with those with hypomorphic genotypes. VACTERL-H alone or in combination with PHENOS is highly associated with FA, but the absence of those features does not rule out the diagnosis of FA.
Keywords: Congenital abnormality; Fanconi anemia; Gene; Genotype-phenotype association; PHENOS; VACTERL-H.
Published by Elsevier Ltd.
Conflict of interest statement
Conflict of Interest
The authors declare no conflicts of interest.
Figures
References
-
- Fanconi G Familiäre infantile perniziosaartige Anämie (pernizioses Blutbild und Konstitution). Jahrbuch für Kinderheilkunde. 1927;117:257–89.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
