

Haploinsufficiency of the Notch Ligand DLL1 Causes Variable Neurodevelopmental Disorders
- PMID: 31353024
- PMCID: PMC6731356
- DOI: 10.1016/j.ajhg.2019.07.002
Haploinsufficiency of the Notch Ligand DLL1 Causes Variable Neurodevelopmental Disorders
Abstract
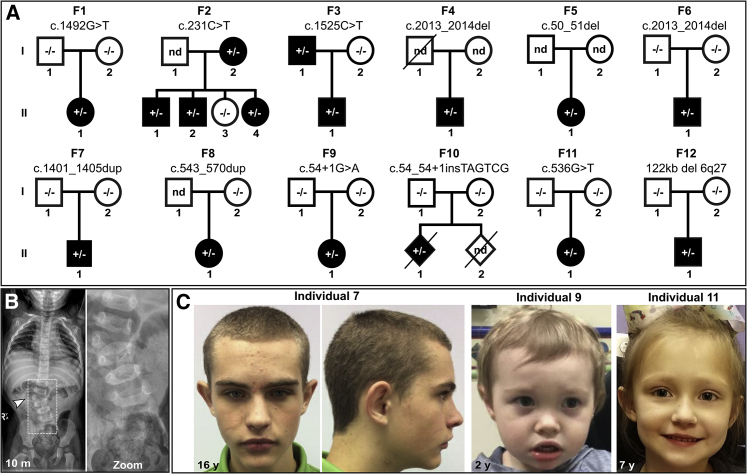
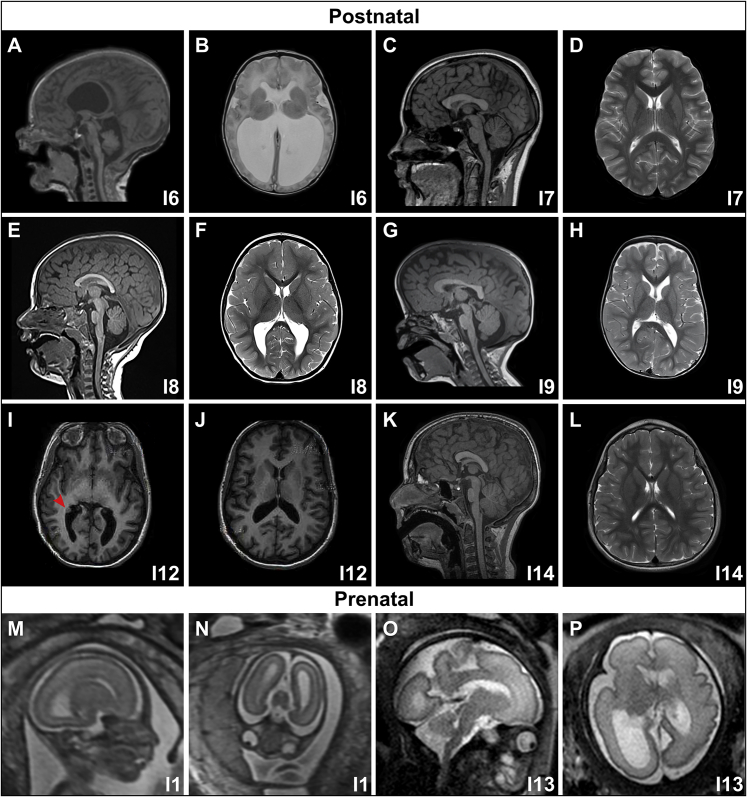
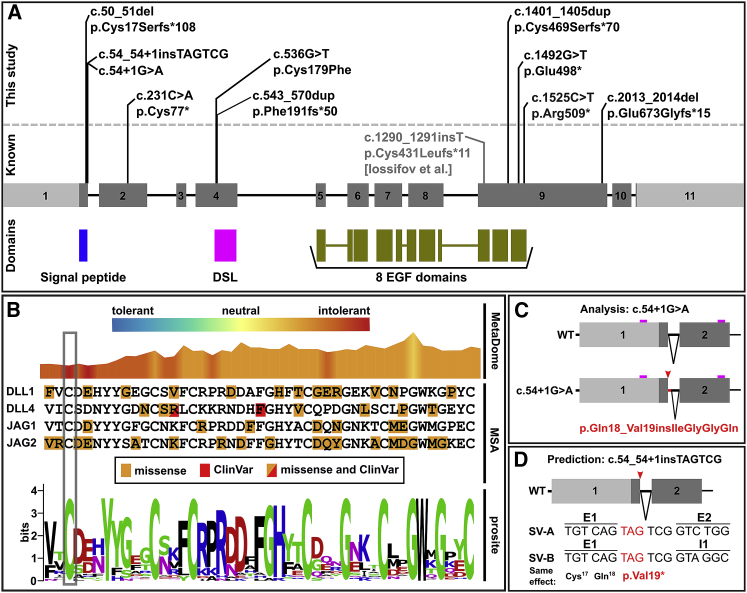
Notch signaling is an established developmental pathway for brain morphogenesis. Given that Delta-like 1 (DLL1) is a ligand for the Notch receptor and that a few individuals with developmental delay, intellectual disability, and brain malformations have microdeletions encompassing DLL1, we hypothesized that insufficiency of DLL1 causes a human neurodevelopmental disorder. We performed exome sequencing in individuals with neurodevelopmental disorders. The cohort was identified using known Matchmaker Exchange nodes such as GeneMatcher. This method identified 15 individuals from 12 unrelated families with heterozygous pathogenic DLL1 variants (nonsense, missense, splice site, and one whole gene deletion). The most common features in our cohort were intellectual disability, autism spectrum disorder, seizures, variable brain malformations, muscular hypotonia, and scoliosis. We did not identify an obvious genotype-phenotype correlation. Analysis of one splice site variant showed an in-frame insertion of 12 bp. In conclusion, heterozygous DLL1 pathogenic variants cause a variable neurodevelopmental phenotype and multi-systemic features. The clinical and molecular data support haploinsufficiency as a mechanism for the pathogenesis of this DLL1-related disorder and affirm the importance of DLL1 in human brain development.
Keywords: DLL1; Notch signaling; autism; brain malformation; developmental delay; intellectual disability; vertebral segmentation defects.
Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
E.T., A.T., Y.S., Y.C., S.L., K.M., and X.W. are employees of GeneDx, Inc., a wholly owned subsidiary of OPKO Health, Inc. All other authors declare no competing interests.
Figures
References
-
- Bray S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016;17:722–735. - PubMed
-
- Louvi A., Artavanis-Tsakonas S. Notch signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006;7:93–102. - PubMed
-
- Bettenhausen B., Hrabĕ de Angelis M., Simon D., Guénet J.L., Gossler A. Transient and restricted expression during mouse embryogenesis of Dll1, a murine gene closely related to Drosophila Delta. Development. 1995;121:2407–2418. - PubMed
-
- Hrabĕ de Angelis M., McIntyre J., 2nd, Gossler A. Maintenance of somite borders in mice requires the Delta homologue DII1. Nature. 1997;386:717–721. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
