

Phenome-wide Burden of Copy-Number Variation in the UK Biobank
- PMID: 31353025
- PMCID: PMC6699064
- DOI: 10.1016/j.ajhg.2019.07.001
Phenome-wide Burden of Copy-Number Variation in the UK Biobank
Abstract
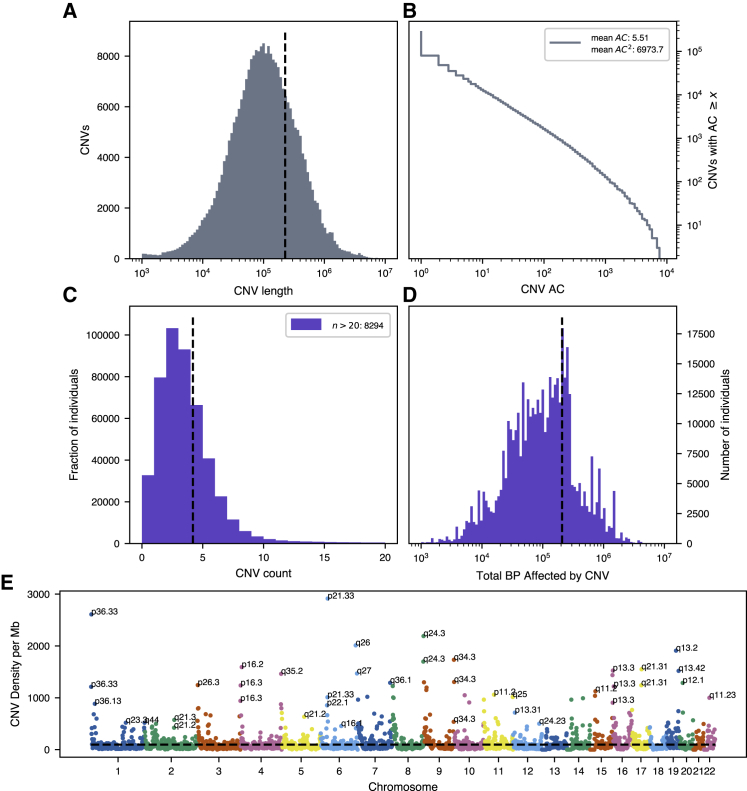
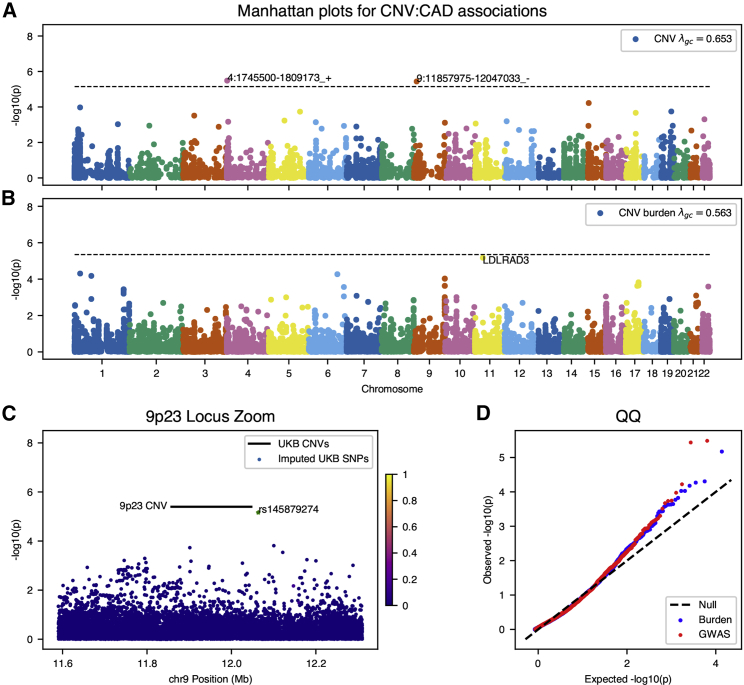
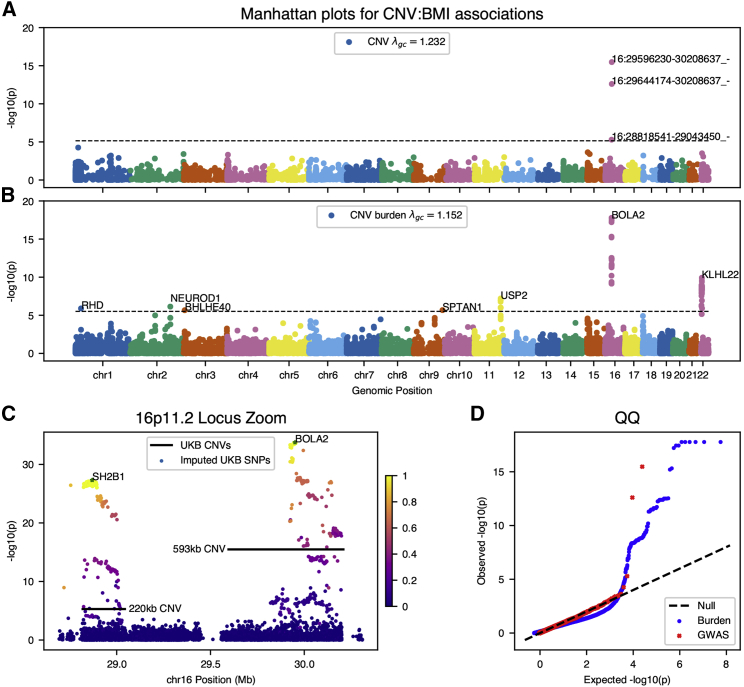
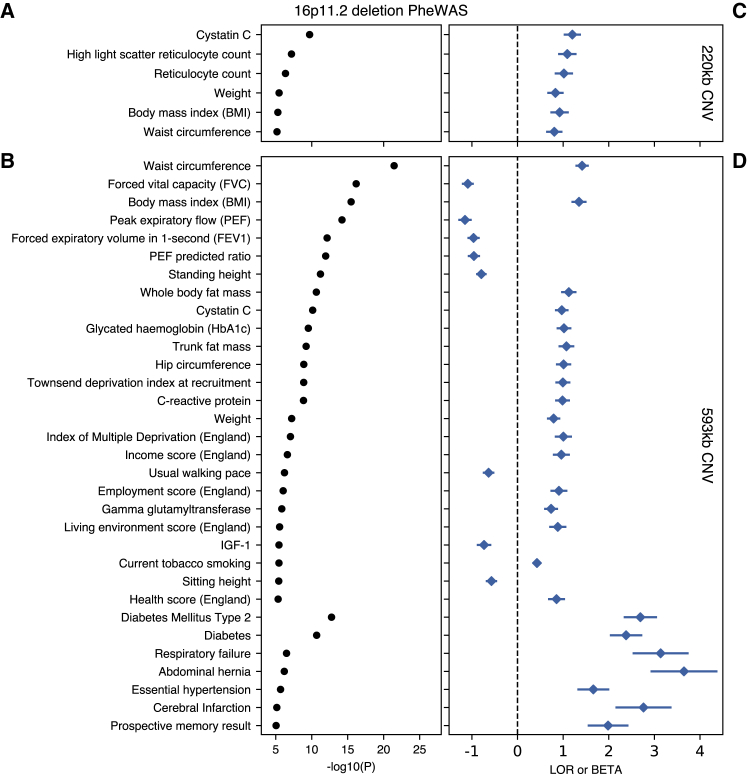
Copy-number variations (CNVs) represent a significant proportion of the genetic differences between individuals and many CNVs associate causally with syndromic disease and clinical outcomes. Here, we characterize the landscape of copy-number variation and their phenome-wide effects in a sample of 472,228 array-genotyped individuals from the UK Biobank. In addition to population-level selection effects against genic loci conferring high mortality, we describe genetic burden from potentially pathogenic and previously uncharacterized CNV loci across more than 3,000 quantitative and dichotomous traits, with separate analyses for common and rare classes of variation. Specifically, we highlight the effects of CNVs at two well-known syndromic loci 16p11.2 and 22q11.2, previously uncharacterized variation at 9p23, and several genic associations in the context of acute coronary artery disease and high body mass index. Our data constitute a deeply contextualized portrait of population-wide burden of copy-number variation, as well as a series of dosage-mediated genic associations across the medical phenome.
Keywords: UK Biobank; association testing; copy number variation; genetics; genomics; microdeletion/microduplication syndrome; population database; selection bias; structural variation.
Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Mikhail F.M. Copy number variations and human genetic disease. Curr. Opin. Pediatr. 2014;26:646–652. - PubMed
-
- Carvill G.L., Mefford H.C. Microdeletion syndromes. Curr. Opin. Genet. Dev. 2013;23:232–239. - PubMed
-
- Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
