

Near full genome characterization of HIV-1 unique recombinant forms in Cameroon reveals dominant CRF02_AG and F2 recombination patterns
- PMID: 31353798
- PMCID: PMC6661401
- DOI: 10.1002/jia2.25362
Near full genome characterization of HIV-1 unique recombinant forms in Cameroon reveals dominant CRF02_AG and F2 recombination patterns
Abstract
Introduction: In Cameroon, a manifold diversity of HIV strains exists with CRF02_AG and unique recombinant forms (URFs) being the predominant strains. In recent years, a steady increase in URFs and clade F2 viruses has been monitored through partial genome sequencing. There is an information gap in the characterization of emerging URFs along the full genome, which is needed to address the challenges URFs pose towards diagnosis, treatment and HIV-1 vaccine design.
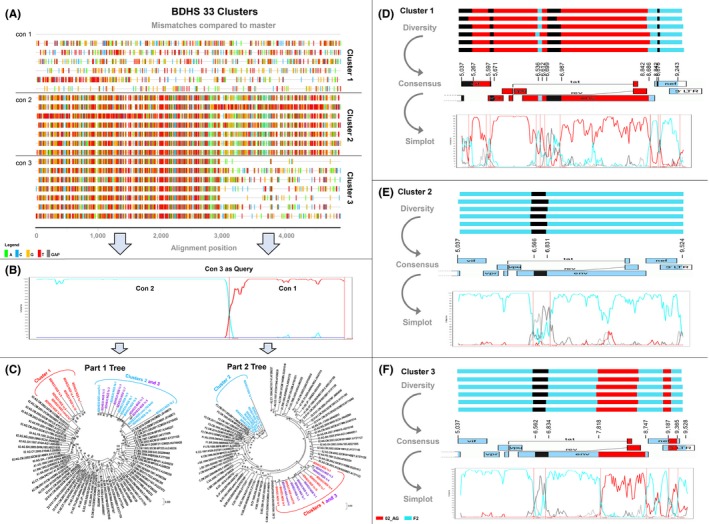
Method: Eighteen Cameroonian URFs from samples collected between the years 2000 and 2015 were studied using a newly developed near full genome sequencing (NFGS) protocol based on variable nested RT-PCRs with a versatile primer set. Near full genomes were characterized for recombination patterns and sequence signatures with possible impact on antiretroviral treatment or Env-directed immune responses. Third-generation sequencing (3GS) of near full or half genomes (HGs) gave insight into intra-patient URF diversity.
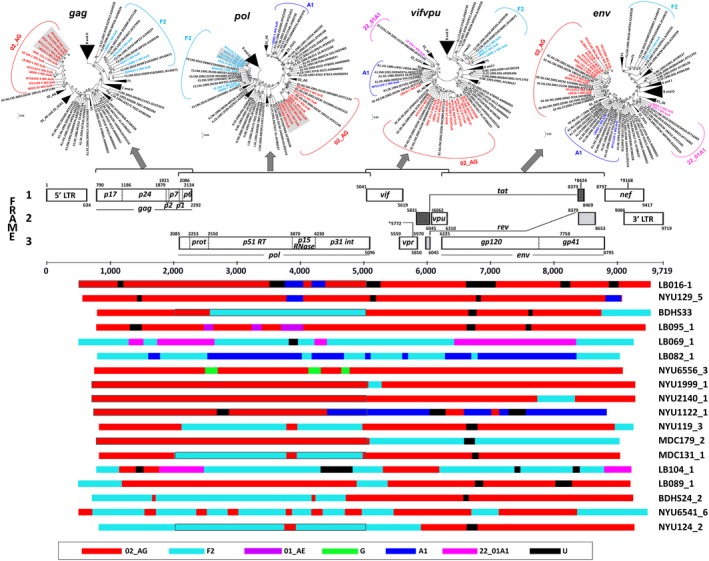
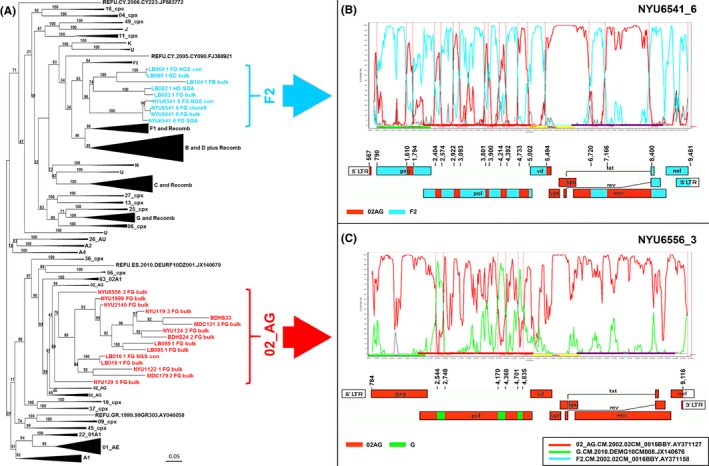
Results: The characterized URFs were composed of a broad variety of subtypes and recombinants including A, F, G, CRF01_AE, CRF02_AG and CRF22_01A1. Phylogenetic analysis unveiled dominant CRF02_AG and F2 recombination patterns. 3GS indicated a high intra-patient URF diversity with up to four distinct viral sub-populations present in plasma at the same time. URF pol genomic analysis revealed a number of accessory drug resistance mutations (DRMs) in the ART-naïve participants. Genotypic env analysis suggests CCR5 usage in 14/18 samples and identified deviations at residues, critical for gp120/gp41 interphase and CD4 binding site broadly neutralizing antibodies in more than half of the studied URFs. V1V2 sites of immune pressure in the human RV144 vaccine study varied in more than a third of URFs.
Conclusions: This study identified novel mosaic patterns in URFs in Cameroon. In line with the regional predominance of CRF_02AG and the increased prevalence of clade F2, prominent CRF_02AG and F2 background patterns were observed underlying the URFs. In the context of the novel mosaic genomes, the impact of the identified accessory DRMs and Env epitope variations on treatment and immune control remains elusive. The evolving diversity of HIV-1 URFs in Cameroon requires continuous monitoring to respond to the increasing challenges for diagnosis, antiretroviral treatment and prevention.
Keywords: Env epitopes and drug resistance mutations; intra-patient viral diversity; near full genome sequencing; third-generation sequencing; unique recombinant forms.
© 2019 The Authors. Journal of the International AIDS Society published by John Wiley & Sons Ltd on behalf of the International AIDS Society.
Figures
References
-
- Hemelaar J, Elangovan R, Yun J, Dickson‐Tetteh L, Fleminger I, Kirtley S, et al. Global and regional molecular epidemiology of HIV‐1, 1990‐2015: a systematic review, global survey, and trend analysis. Lancet Infect Dis. 2019;19(2):143–55. - PubMed
-
- McNeil D Jr. Chimp to man to history: the path of AIDS. The New York Times. 2011 October 17, 2011. [cited 2018 Nov 19]. Available from: https://www.nytimes.com/2011/10/18/health/18aids.html
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
