

Can Targeting Non-Contiguous V-Regions With Paired-End Sequencing Improve 16S rRNA-Based Taxonomic Resolution of Microbiomes?: An In Silico Evaluation
- PMID: 31354793
- PMCID: PMC6640118
- DOI: 10.3389/fgene.2019.00653
Can Targeting Non-Contiguous V-Regions With Paired-End Sequencing Improve 16S rRNA-Based Taxonomic Resolution of Microbiomes?: An In Silico Evaluation
Abstract
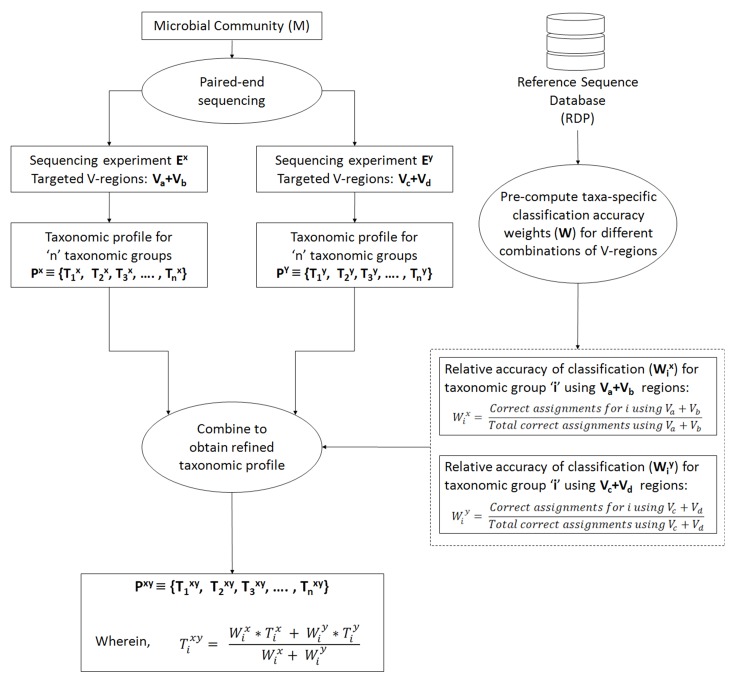
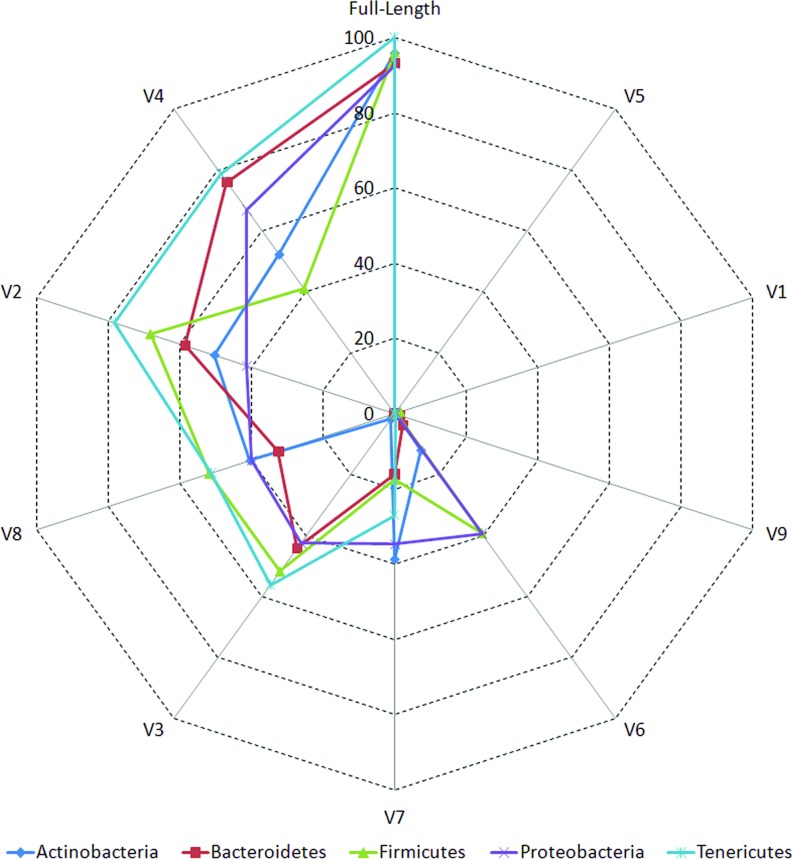
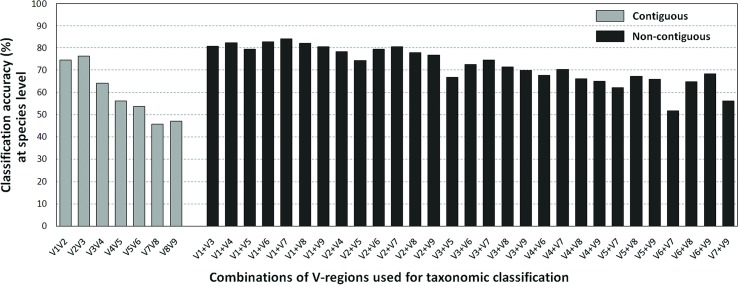
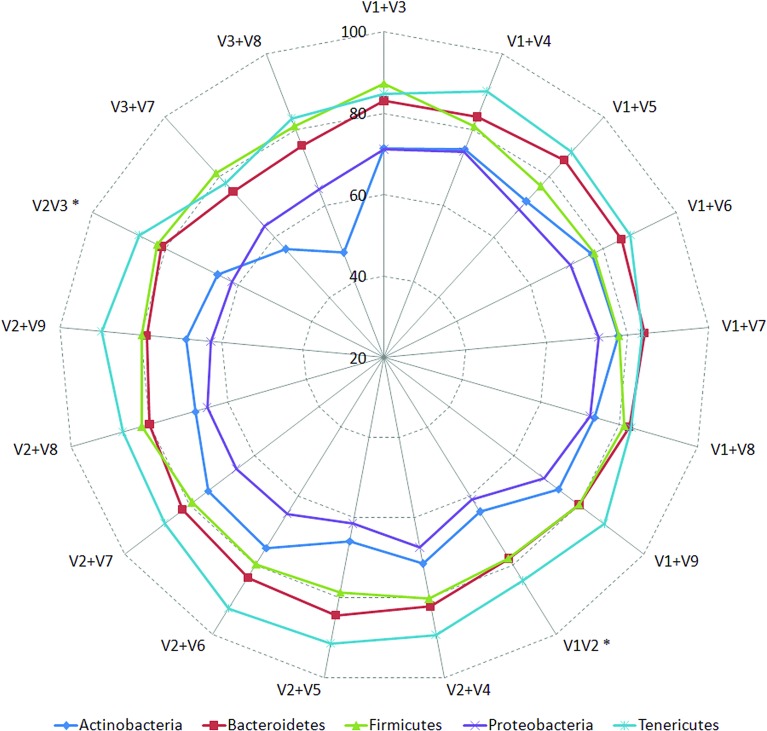
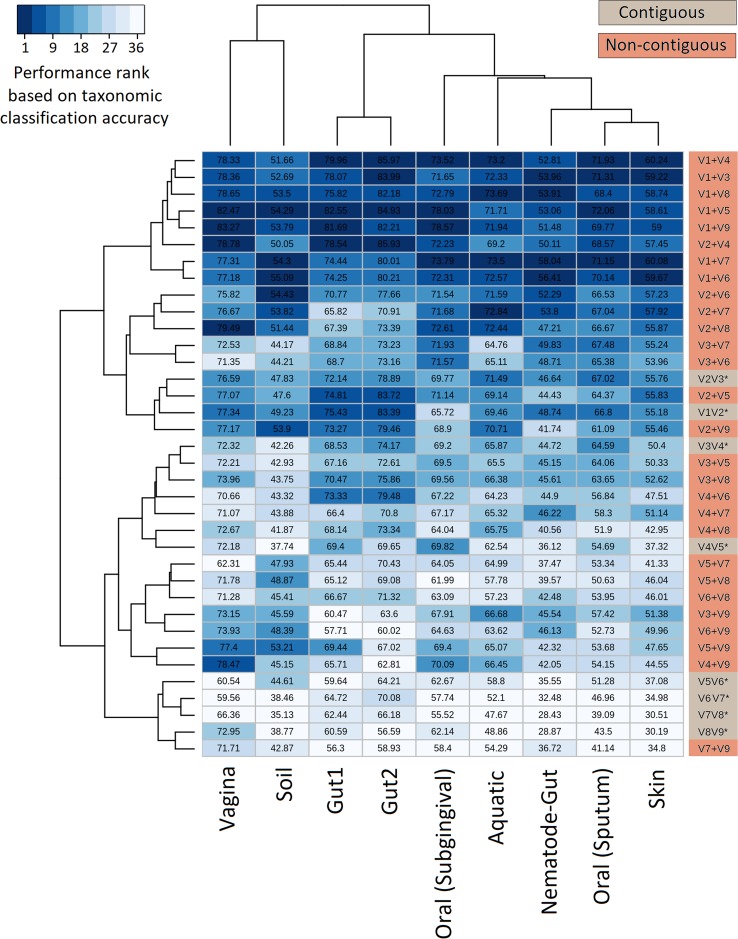
Background: Next-generation sequencing (NGS) technologies have enabled probing of microbial diversity in different environmental niches with unprecedented sequencing depth. However, due to read-length limitations of popular NGS technologies, 16S amplicon sequencing-based microbiome studies rely on targeting short stretches of the 16S rRNA gene encompassing a selection of variable (V) regions. In most cases, such a short stretch constitutes a single V-region or a couple of V-regions placed adjacent to each other on the 16S rRNA gene. Given that different V-regions have different resolving ability with respect to various taxonomic groups, selecting the optimal V-region (or a combination thereof) remains a challenge. Methods: The accuracy of taxonomic profiles generated from sequences encompassing 1) individual V-regions, 2) adjacent V-regions, and 3) pairs of non-contiguous V-regions were assessed and compared. Subsequently, the discriminating capability of different V-regions with respect to different taxonomic lineages was assessed. The possibility of using paired-end sequencing protocols to target combinations of non-adjacent V-regions was finally evaluated with respect to the utility of such an experimental design in providing improved taxonomic resolution. Results: Extensive validation with simulated microbiome datasets mimicking different environmental and host-associated microbiome samples suggest that targeting certain combinations of non-contiguously placed V-regions might yield better taxonomic classification accuracy compared to conventional 16S amplicon sequencing targets. This work also puts forward a novel in silico combinatorial strategy that enables creation of consensus taxonomic profiles from experiments targeting multiple pair-wise combinations of V-regions to improve accuracy in taxonomic classification. Conclusion: The study suggests that targeting non-contiguous V-regions with paired-end sequencing can improve 16S rRNA-based taxonomic resolution of microbiomes. Furthermore, employing the novel in silico combinatorial strategy can improve taxonomic classification without any significant additional experimental costs and/or efforts. The empirical observations obtained can potentially serve as a guideline for future 16S microbiome studies, and facilitate researchers in choosing the optimal combination of V-regions for a specific experiment/sampled environment.
Keywords: amplicon sequencing; metagenomics 16S; microbiome analysis; paired-end sequencing; taxonomic profiling.
Figures
References
-
- Bartram A. K., Lynch M. D. J., Stearns J. C., Moreno-Hagelsieb G., Neufeld J. D. (2011). Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end illumina reads. Appl. Environ. Microbiol. 77, 3846–3852. 10.1128/AEM.02772-10 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases