

Complex patterns of inheritance, including synergistic heterozygosity, in inborn errors of metabolism: Implications for precision medicine driven diagnosis and treatment
- PMID: 31358473
- PMCID: PMC8931500
- DOI: 10.1016/j.ymgme.2019.07.011
Complex patterns of inheritance, including synergistic heterozygosity, in inborn errors of metabolism: Implications for precision medicine driven diagnosis and treatment
Abstract
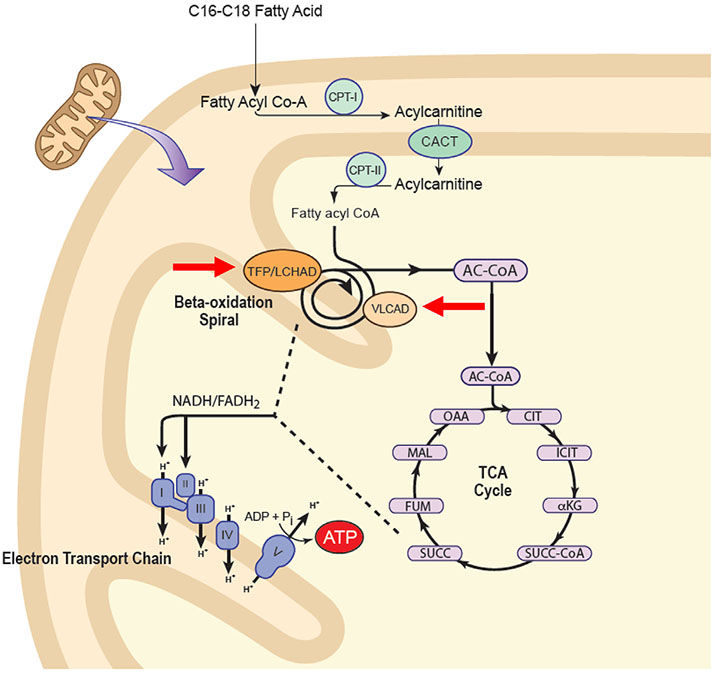
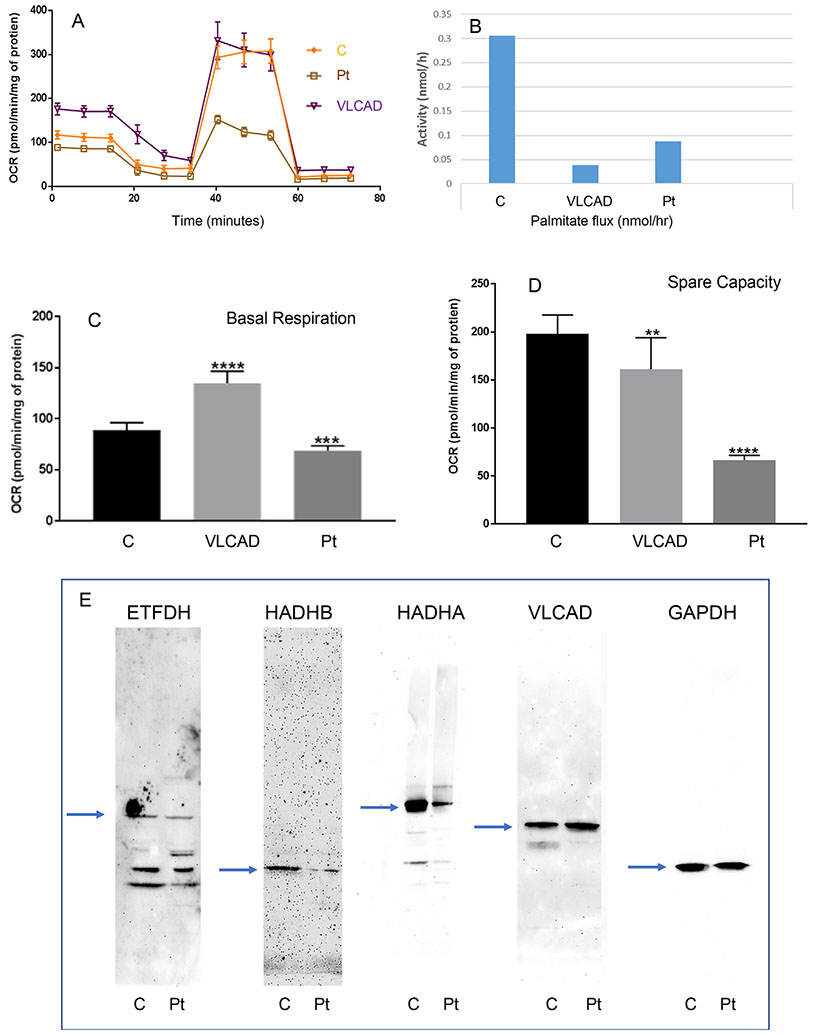
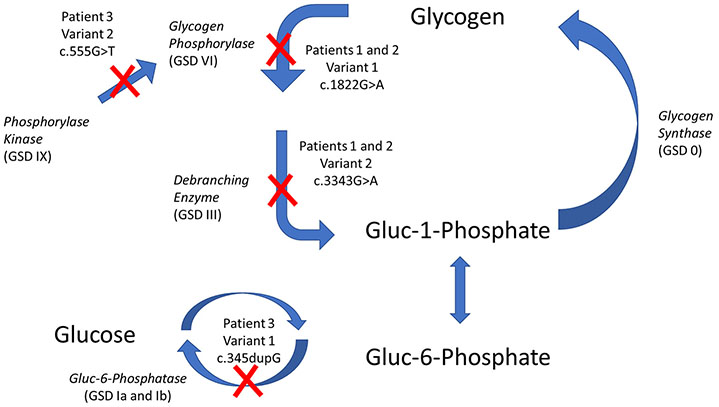
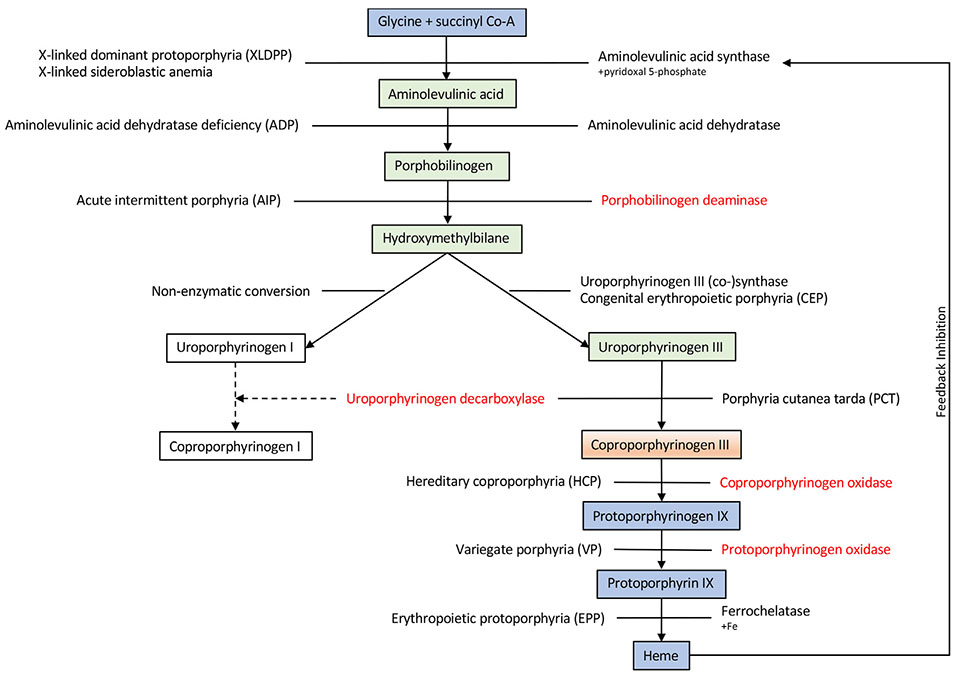
Inborn errors of metabolism have traditionally been viewed as the quintessential single gene disorders; defects in one gene leads to loss of activity of one enzyme causing a metabolic imbalance and clinical disease. However, reality has never been quite that simple, and the classic "one gene-one enzyme" paradigm has been upended in many ways. Multiple gene defects can lead to the same biochemical phenotype, often with different clinical symptoms. Additionally, different mutations in the same gene can cause variable phenotypes, often most dramatic when a disease can be identified by pre-symptomatic screening. Moreover, response to therapy is not homogeneous across diseases and specific mutations. Perhaps the biggest deviation from traditional monogenic inheritance is in the setting of synergistic heterozygosity, a multigenic inheritance pattern in which mutations in multiple genes in a metabolic pathway lead to sufficient disruption of flux through the pathway, mimicking a monogenic disorder caused by homozygous defects in one gene in that pathway. In addition, widespread adoption of whole exome and whole genome sequencing in medical genetics has led to the realization that individual patients with apparently hybrid phenotypes can have mutations in more than one gene, leading to a mixed genetic disorder. Each of these situations point to a need for as much precision as possible in diagnosing metabolic disease, and it is likely to become increasingly critical to drive therapy. This article examines examples in traditional monogenic disorders that illustrates these points and define inborn errors of metabolism as complex genetic traits on the leading edge of precision medicine.
Copyright © 2019. Published by Elsevier Inc.
Figures
References
-
- Grosse SD, Dezateux C, Newborn screening for inherited metabolic disease, Lancet 369 (2007) 5–6. - PubMed
-
- Rinaldo P, Lim JS, Tortorelli S, Gavrilov D, Matern D, Newborn screening of metabolic disorders: recent progress and future developments, Nestle Nutr Workshop Ser Pediatr Program 62 (2008) 81–93 (discussion 93-86). - PubMed
-
- Bennett MJ, Rinaldo P, Wilcken B, Pass KA, Watson MS, Wanders RJ, Newborn screening for metabolic disorders: how are we doing, and where are we going? Clin. Chem 58 (2012) 324–331. - PubMed
-
- Wasserstein MP, Caggana M, Bailey SM, Desnick RJ, Edelmann L, Estrella L, Holzman I, Kelly NR, Kornreich R, Kupchik SG, Martin M, Nafday SM, Wasserman R, Yang A, Yu C, Orsini JJ, The New York pilot newborn screening program for lysosomal storage diseases: report of the first 65,000 infants, Genet Med 21 (2019) 631–640. - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
