

Inhibition of TGF- β 1 Signaling by IL-15: A Novel Role for IL-15 in the Control of Renal Epithelial-Mesenchymal Transition: IL-15 Counteracts TGF- β 1-Induced EMT in Renal Fibrosis
- PMID: 31360169
- PMCID: PMC6642769
- DOI: 10.1155/2019/9151394
Inhibition of TGF- β 1 Signaling by IL-15: A Novel Role for IL-15 in the Control of Renal Epithelial-Mesenchymal Transition: IL-15 Counteracts TGF- β 1-Induced EMT in Renal Fibrosis
Abstract
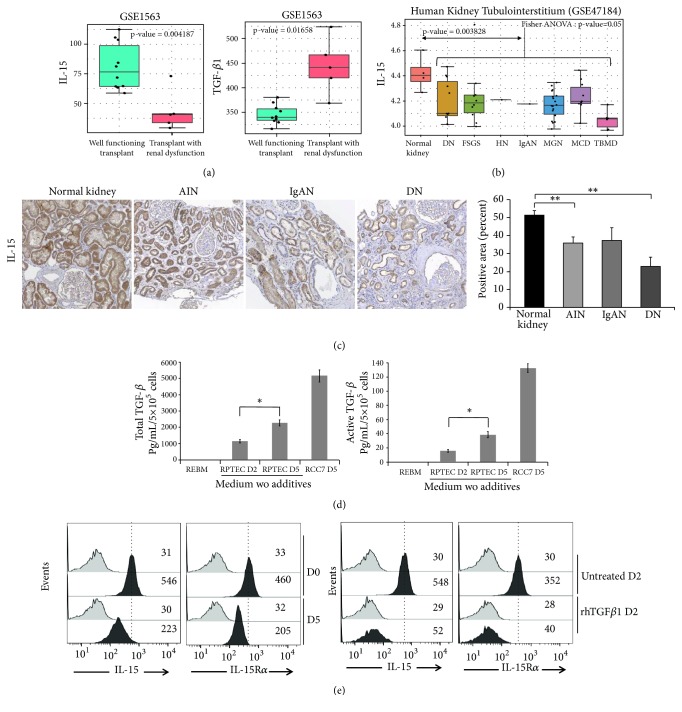
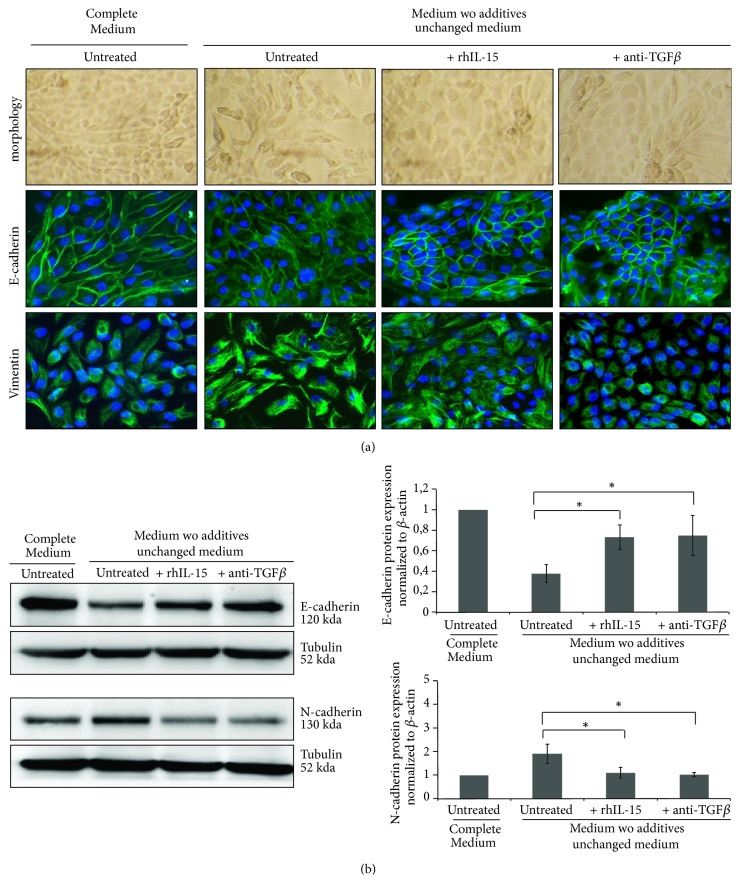
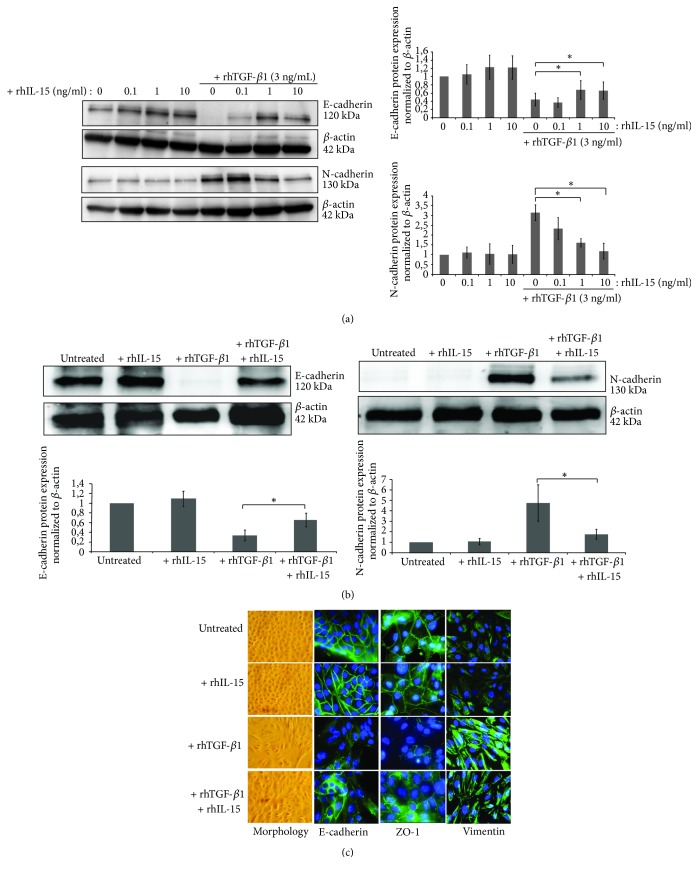
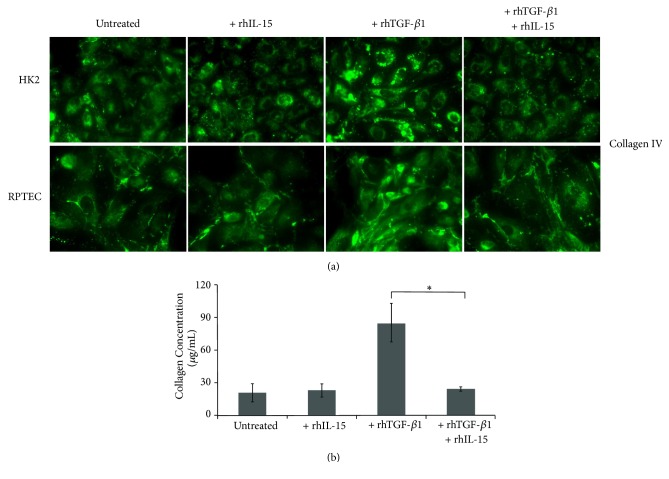
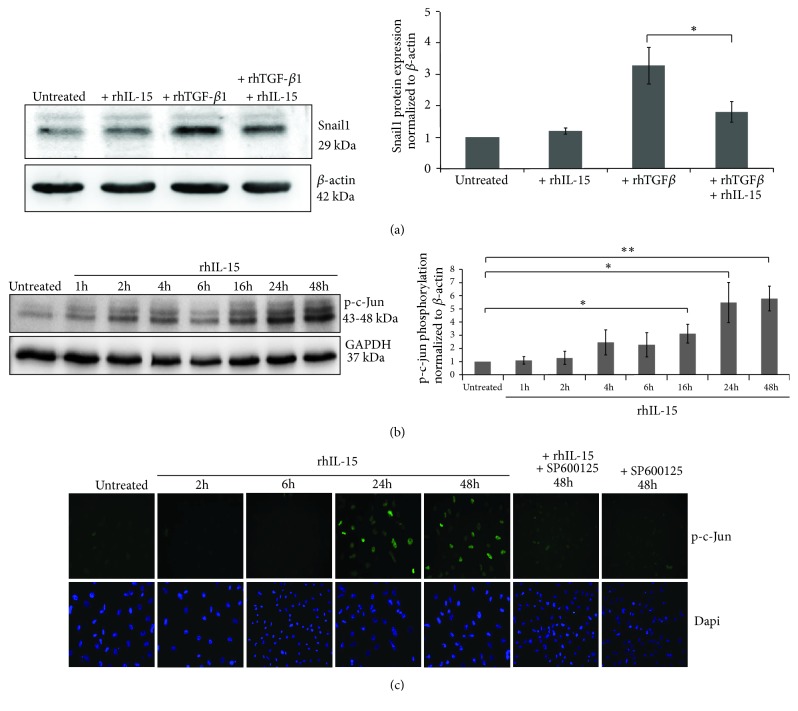
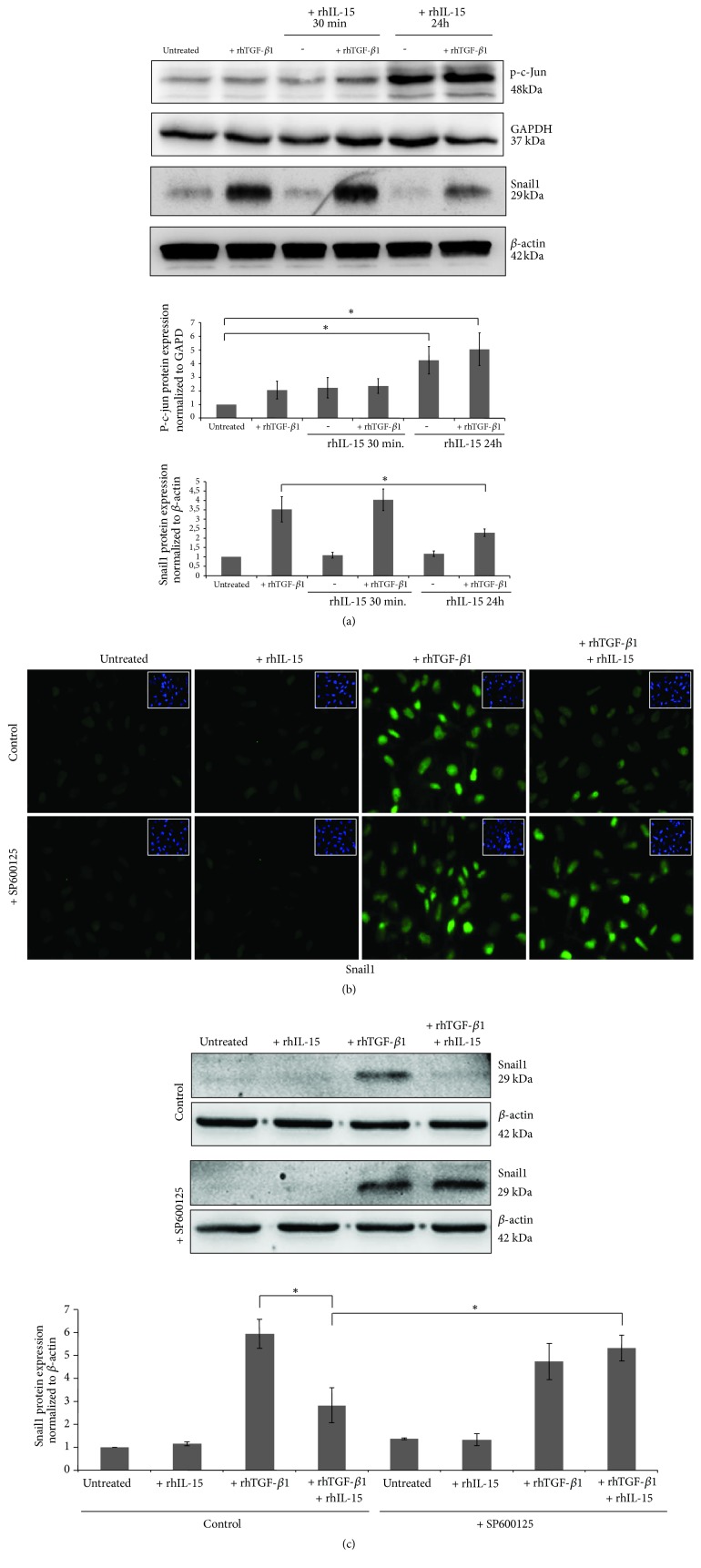
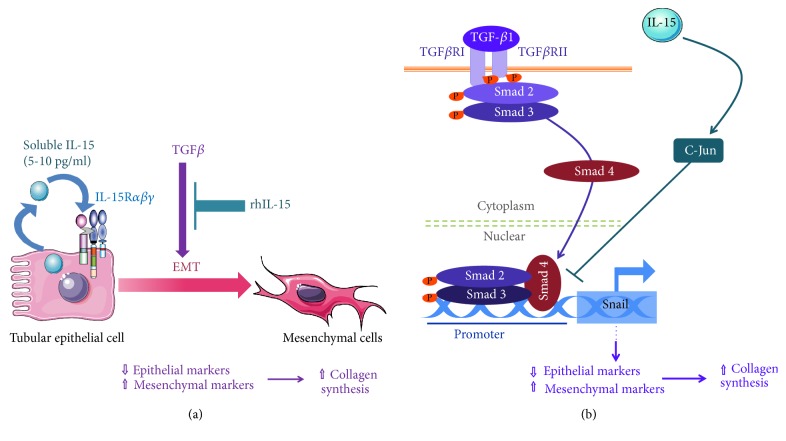
Renal tubulointerstitial fibrosis is the final common pathway in end-stage renal disease and is characterized by aberrant accumulation of extracellular matrix (ECM) components secreted by myofibroblasts. Tubular type 2 EMT, induced by TGF-β, plays an important role in renal fibrosis, by participating directly or indirectly in myofibroblasts generation. TGF-β1-induced apoptosis and fibrosis in experimental chronic murine kidney diseases are concomitantly associated with an intrarenal decreased expression of the IL-15 survival factor. Since IL-15 counteracts TGF-β1 effects in different cell models, we analyzed whether (1) human chronic inflammatory nephropathies evolving towards fibrosis could be also characterized by a weak intrarenal IL-15 expression and (2) IL-15 could inhibit epithelial-mesenchymal transition (EMT) and excess matrix deposition in human renal proximal tubular epithelial cells (RPTEC). Our data show that different human chronic kidney diseases are characterized by a strong decreased expression of intrarenal IL-15, which is particularly relevant in diabetic nephropathy, in which type 2 tubular EMT plays an important role in fibrosis. Moreover, primary epithelial tubular cultures deprived of growth supplements rapidly produce active TGF-β1 inducing a "spontaneous" EMT process characterized by the loss of membrane-bound IL-15 (mbIL-15) expression. Both "spontaneous" EMT and recombinant human (rh) TGF-β1-induced EMT models can be inhibited by treating RPTEC and HK2 cells with rhIL-15. Through a long-lasting phospho-c-jun activation, IL-15 inhibits rhTGF-β1-induced Snail1 expression, the master inducer of EMT, and blocks TGF-β1-induced tubular EMT and downstream collagen synthesis. In conclusion, our data suggest that intrarenal IL-15 could be a natural inhibitor of TGF-β in human kidney able to guarantee epithelial homeostasis and to prevent EMT process. Thus, both in vivo and in vitro an unbalance in intrarenal IL-15 and TGF-β1 levels could render RPTEC cells more prone to undergo EMT process. Exogenous IL-15 treatment could be beneficial in some human nephropathies such as diabetic nephropathy.
Figures
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
