

Small RNAs from the plant pathogenic fungus Sclerotinia sclerotiorum highlight host candidate genes associated with quantitative disease resistance
- PMID: 31361080
- PMCID: PMC6715603
- DOI: 10.1111/mpp.12841
Small RNAs from the plant pathogenic fungus Sclerotinia sclerotiorum highlight host candidate genes associated with quantitative disease resistance
Abstract
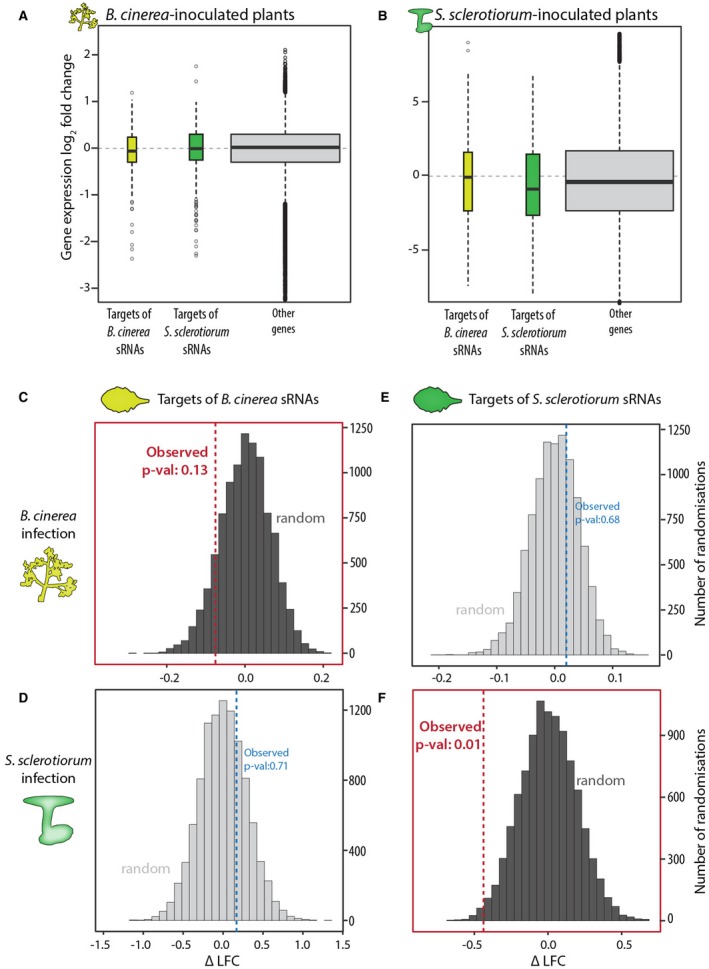
Fungal plant pathogens secrete effector proteins and metabolites to cause disease. Additionally, some species transfer small RNAs (sRNAs) into plant cells to silence host mRNAs through complementary base pairing and suppress plant immunity. The fungus Sclerotinia sclerotiorum infects over 600 plant species, but little is known about the molecular processes that govern interactions with its many hosts. In particular, evidence for the production of sRNAs by S. sclerotiorum during infection is lacking. We sequenced sRNAs produced by S. sclerotiorum in vitro and during infection of two host species, Arabidopsis thaliana and Phaseolus vulgaris. We found that S. sclerotiorum produces at least 374 distinct highly abundant sRNAs during infection, mostly originating from repeat-rich plastic genomic regions. We predicted the targets of these sRNAs in A. thaliana and found that these genes were significantly more down-regulated during infection than the rest of the genome. Predicted targets of S. sclerotiorum sRNAs in A. thaliana were enriched for functional domains associated with plant immunity and were more strongly associated with quantitative disease resistance in a genome-wide association study (GWAS) than the rest of the genome. Mutants in A. thaliana predicted sRNA target genes SERK2 and SNAK2 were more susceptible to S. sclerotiorum than wild-type, suggesting that S. sclerotiorum sRNAs may contribute to the silencing of immune components in plants. The prediction of fungal sRNA targets in plant genomes can be combined with other global approaches, such as GWAS, to assist in the identification of plant genes involved in quantitative disease resistance.
Keywords: Arabidopsis; GWAS; RNAi; effector; necrotrophic fungus; plant immunity; plant pathogen.
© 2019 The Authors. Molecular Plant Pathology published by British Society for Plant Pathology and John Wiley & Sons Ltd.
Figures






Similar articles
-
Genome-wide identification of Sclerotinia sclerotiorum small RNAs and their endogenous targets.BMC Genomics. 2023 Oct 2;24(1):582. doi: 10.1186/s12864-023-09686-7. BMC Genomics. 2023. PMID: 37784009 Free PMC article.
-
Rapid identification of an Arabidopsis NLR gene as a candidate conferring susceptibility to Sclerotinia sclerotiorum using time-resolved automated phenotyping.Plant J. 2020 Jul;103(2):903-917. doi: 10.1111/tpj.14747. Epub 2020 Apr 21. Plant J. 2020. PMID: 32170798 Free PMC article.
-
Expression polymorphism at the ARPC4 locus links the actin cytoskeleton with quantitative disease resistance to Sclerotinia sclerotiorum in Arabidopsis thaliana.New Phytol. 2019 Apr;222(1):480-496. doi: 10.1111/nph.15580. Epub 2018 Dec 3. New Phytol. 2019. PMID: 30393937
-
Trans-Kingdom sRNA Silencing in Sclerotinia sclerotiorum for Crop Fungal Disease Management.Pathogens. 2025 Apr 21;14(4):398. doi: 10.3390/pathogens14040398. Pathogens. 2025. PMID: 40333207 Free PMC article. Review.
-
The diversity, biogenesis, and activities of endogenous silencing small RNAs in Arabidopsis.Annu Rev Plant Biol. 2014;65:473-503. doi: 10.1146/annurev-arplant-050213-035728. Epub 2014 Feb 26. Annu Rev Plant Biol. 2014. PMID: 24579988 Review.
Cited by
-
Cross-kingdom small RNA communication between plants and fungal phytopathogens-recent updates and prospects for future agriculture.RNA Biol. 2023 Jan;20(1):109-119. doi: 10.1080/15476286.2023.2195731. RNA Biol. 2023. PMID: 36988190 Free PMC article. Review.
-
Polygenic strategies for host-specific and general virulence of Botrytis cinerea across diverse eudicot hosts.Genetics. 2025 Jul 9;230(3):iyaf079. doi: 10.1093/genetics/iyaf079. Genetics. 2025. PMID: 40488422 Free PMC article.
-
Host induced gene silencing of the Sclerotinia sclerotiorum ABHYDROLASE-3 gene reduces disease severity in Brassica napus.PLoS One. 2022 Aug 26;17(8):e0261102. doi: 10.1371/journal.pone.0261102. eCollection 2022. PLoS One. 2022. PMID: 36018839 Free PMC article.
-
Genome-wide identification of Sclerotinia sclerotiorum small RNAs and their endogenous targets.BMC Genomics. 2023 Oct 2;24(1):582. doi: 10.1186/s12864-023-09686-7. BMC Genomics. 2023. PMID: 37784009 Free PMC article.
-
Sclerotinia sclerotiorum Thioredoxin1 (SsTrx1) is required for pathogenicity and oxidative stress tolerance.Mol Plant Pathol. 2021 Nov;22(11):1413-1426. doi: 10.1111/mpp.13127. Epub 2021 Aug 30. Mol Plant Pathol. 2021. PMID: 34459563 Free PMC article.
References
-
- Alexa, A. , Rahnenführer, J. and Lengauer, T. (2006) Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics, 22, 1600–1607. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
