

Revising rapid-onset dystonia-parkinsonism: Broadening indications for ATP1A3 testing
- PMID: 31361359
- PMCID: PMC6879786
- DOI: 10.1002/mds.27801
Revising rapid-onset dystonia-parkinsonism: Broadening indications for ATP1A3 testing
Abstract
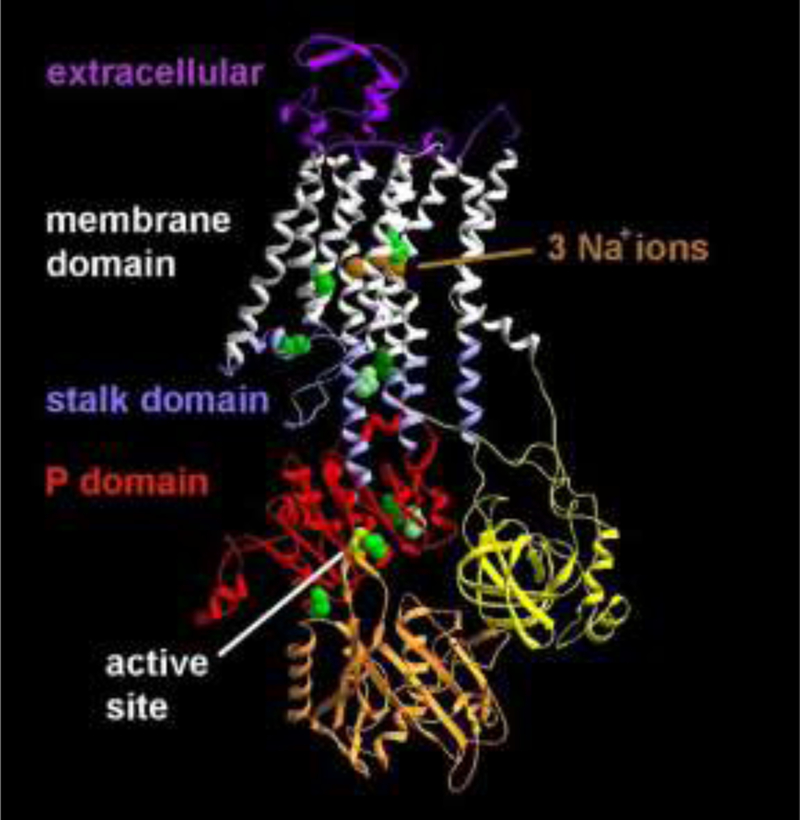
Background and objectives: Rapid-onset dystonia-parkinsonism (RDP) is caused by mutations in the ATP1A3 gene, which codes for the α-3 subunit of the Na+ /K+ ATPase. It has been characterized by rapid-onset bulbar dysfunction, limb dystonia, bradykinesia, and a rostrocaudal spatial gradient of expression, usually after a physiologic trigger. We reexamined whether these features were in fact characteristic.
Methods: We characterized phenotypic variation within a cohort of 50 ATP1A3 mutation-positive individuals (carriers) and 44 mutation-negative family members (noncarriers). Potential participants were gathered through referral for clinical suspicion of RDP or alternating hemiplegia of childhood. Inclusion criteria were having a ATP1A3 mutation or being a family member of such an individual.
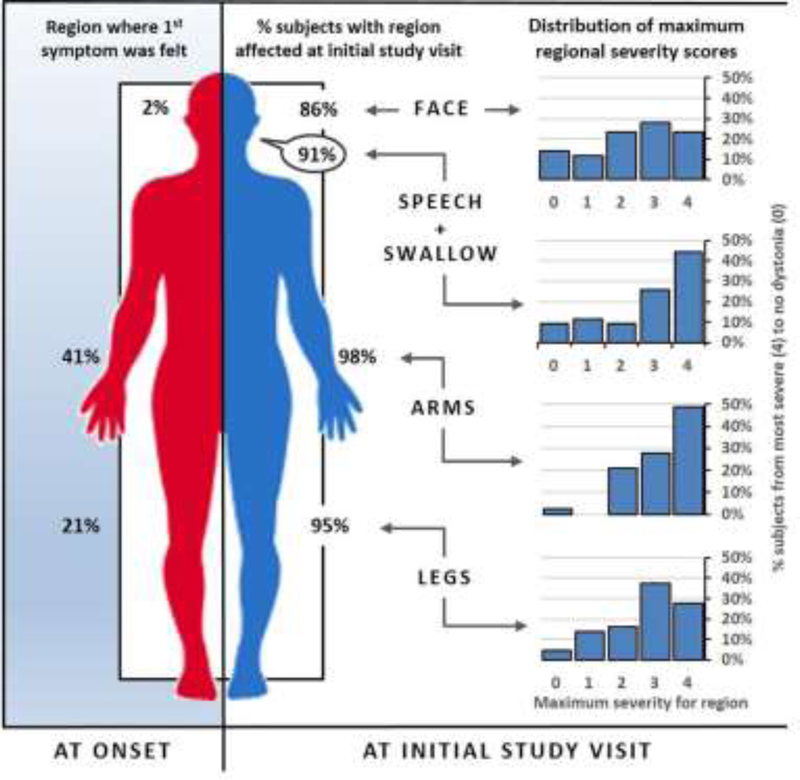
Results: We found RDP is underdiagnosed if only "characteristic" patients are tested. Rapid onset and bulbar predominance were not universally present in carriers. Among those with at least mild symptoms of dystonia, rostrocaudal severity gradient was rare (7%). Symptoms began focally but progressed to be generalized (51%) or multifocal (49%). Arm (41%) onset was most common. Arms and voice were typically most severely affected (48% and 44%, respectively). Triggers preceded onset in 77% of the participants. Rapid onset, dystonia, parkinsonism, bulbar symptoms, headaches, seizures, frontal impairment, and a history of mood disorder and a history of psychosis were more common in carriers. Approximately half of the proband mutations occurred de novo (56%).
Conclusions: Our findings suggest that patients should not be excluded from ATP1A3 testing because of slow onset, limb onset, absent family history, or onset in middle adulthood. RDP should be strongly considered in the differential for any bulbar dystonia. © 2019 International Parkinson and Movement Disorder Society.
© 2019 International Parkinson and Movement Disorder Society.
Conflict of interest statement
Disclosures:
• Dr. Brashear has salary support from NINDS R01NS058949 (this project), performs research for Revance and consults for Revance and Ipsen. Her conflicts of interest are being managed by Wake Forest School of Medicine.
Figures
References
-
- Dobyns WB, Ozelius LJ, Kramer PL, et al. Rapid-onset dystonia-parkinsonism. Neurology 1993;43(12):2596–2602. - PubMed
-
- Brashear A, Dobyns WB, de Carvalho Aguiar P, et al. The phenotypic spectrum of rapid-onset dystonia-parkinsonism (RDP) and mutations in the ATP1A3 gene. Brain 2007;130(Pt 3):828–835. - PubMed
-
- Pittock SJ, Joyce C, O’Keane V, et al. Rapid-onset dystonia-parkinsonism: a clinical and genetic analysis of a new kindred. Neurology 2000;55(7):991–995. - PubMed
-
- de Carvalho Aguiar P, Sweadner KJ, Penniston JT, et al. Mutations in the Na+/K+ -ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 2004;43(2):169–175. - PubMed
-
- Bottger P, Tracz Z, Heuck A, Nissen P, Romero-Ramos M, Lykke-Hartmann K. Distribution of Na/K-ATPase alpha 3 isoform, a sodium-potassium P-type pump associated with rapid-onset of dystonia parkinsonism (RDP) in the adult mouse brain. J Comp Neurol 2011;519(2):376–404. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- R01 NS058949/NS/NINDS NIH HHS/United States
- P50 AA026117/AA/NIAAA NIH HHS/United States
- R01 AG054491/AG/NIA NIH HHS/United States
- R01 NS082453/NS/NINDS NIH HHS/United States
- U24 NS107197/NS/NINDS NIH HHS/United States
- R01 NS091602/NS/NINDS NIH HHS/United States
- R01 AG040282/AG/NIA NIH HHS/United States
- RF1 AG058829/AG/NIA NIH HHS/United States
- P30 AG049638/AG/NIA NIH HHS/United States
- UL1 TR001420/TR/NCATS NIH HHS/United States
- R01 AG055122/AG/NIA NIH HHS/United States
- P01 CA207206/CA/NCI NIH HHS/United States
- R01 MH116675/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials
