

Analyzing Learned Molecular Representations for Property Prediction
- PMID: 31361484
- PMCID: PMC6727618
- DOI: 10.1021/acs.jcim.9b00237
Analyzing Learned Molecular Representations for Property Prediction
Erratum in
-
Correction to Analyzing Learned Molecular Representations for Property Prediction.J Chem Inf Model. 2019 Dec 23;59(12):5304-5305. doi: 10.1021/acs.jcim.9b01076. Epub 2019 Dec 9. J Chem Inf Model. 2019. PMID: 31814400 Free PMC article. No abstract available.
Abstract
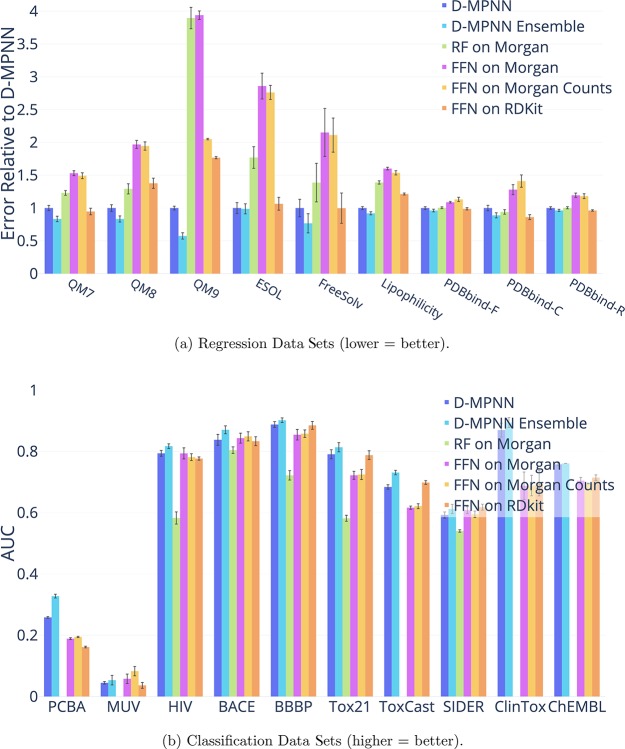
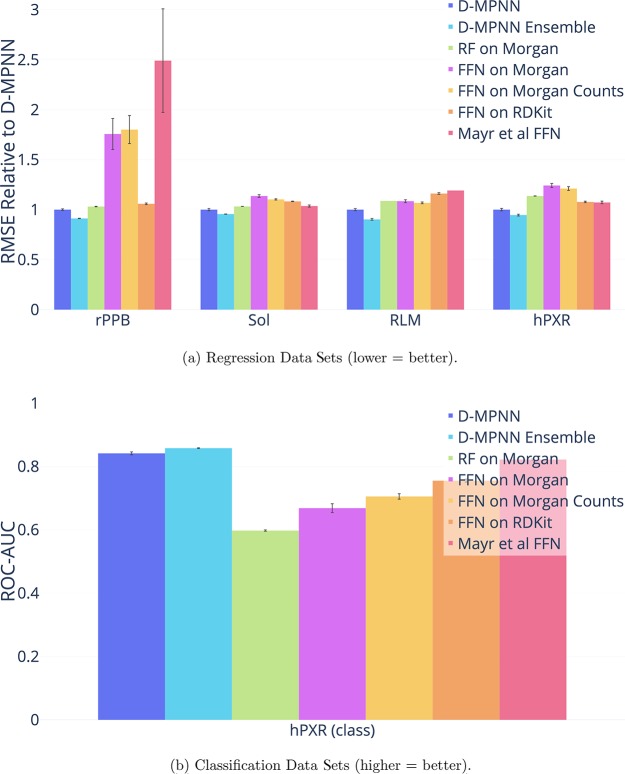
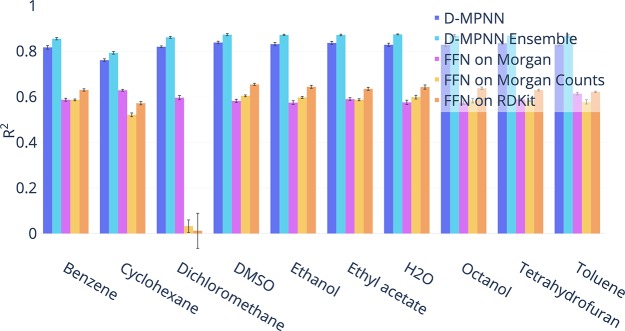
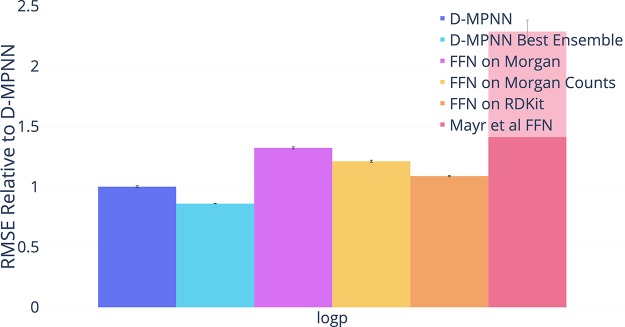
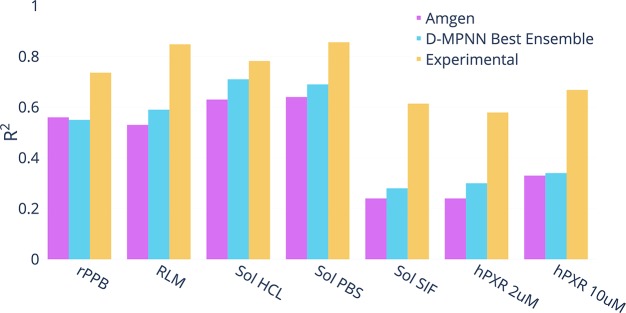
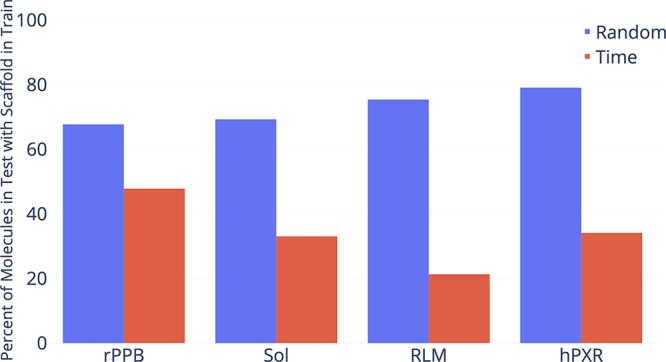
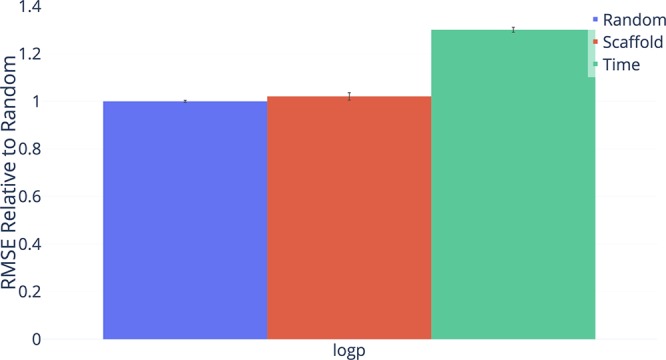
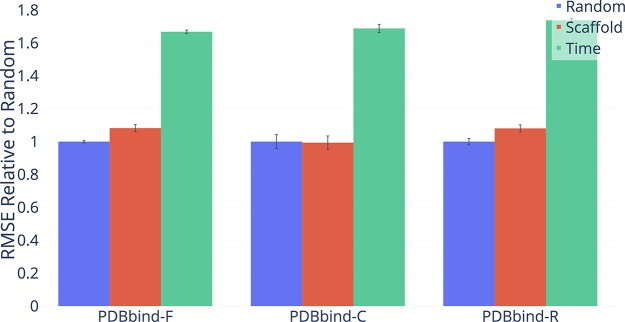
Advancements in neural machinery have led to a wide range of algorithmic solutions for molecular property prediction. Two classes of models in particular have yielded promising results: neural networks applied to computed molecular fingerprints or expert-crafted descriptors and graph convolutional neural networks that construct a learned molecular representation by operating on the graph structure of the molecule. However, recent literature has yet to clearly determine which of these two methods is superior when generalizing to new chemical space. Furthermore, prior research has rarely examined these new models in industry research settings in comparison to existing employed models. In this paper, we benchmark models extensively on 19 public and 16 proprietary industrial data sets spanning a wide variety of chemical end points. In addition, we introduce a graph convolutional model that consistently matches or outperforms models using fixed molecular descriptors as well as previous graph neural architectures on both public and proprietary data sets. Our empirical findings indicate that while approaches based on these representations have yet to reach the level of experimental reproducibility, our proposed model nevertheless offers significant improvements over models currently used in industrial workflows.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
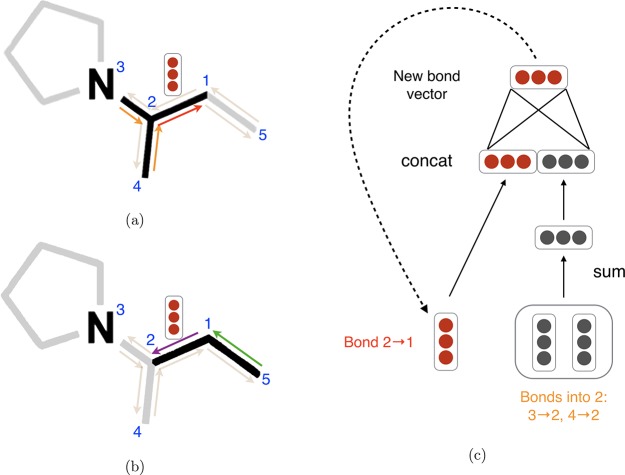
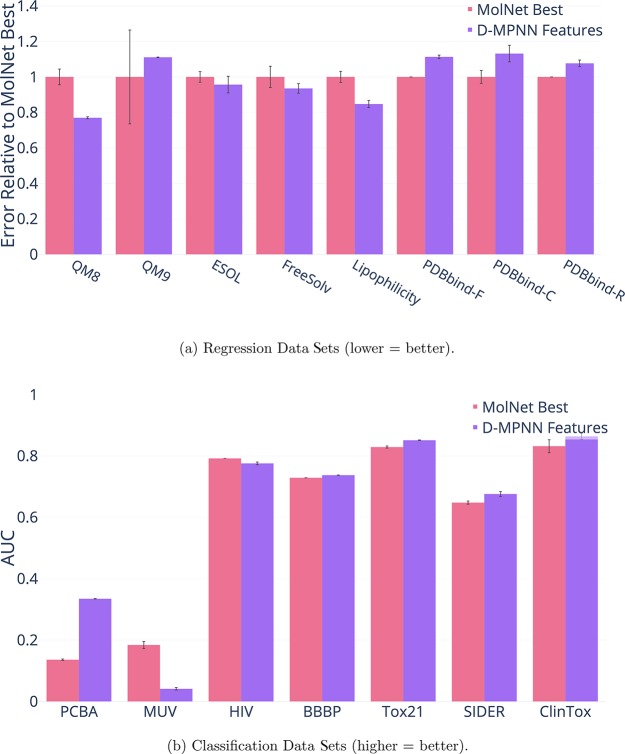
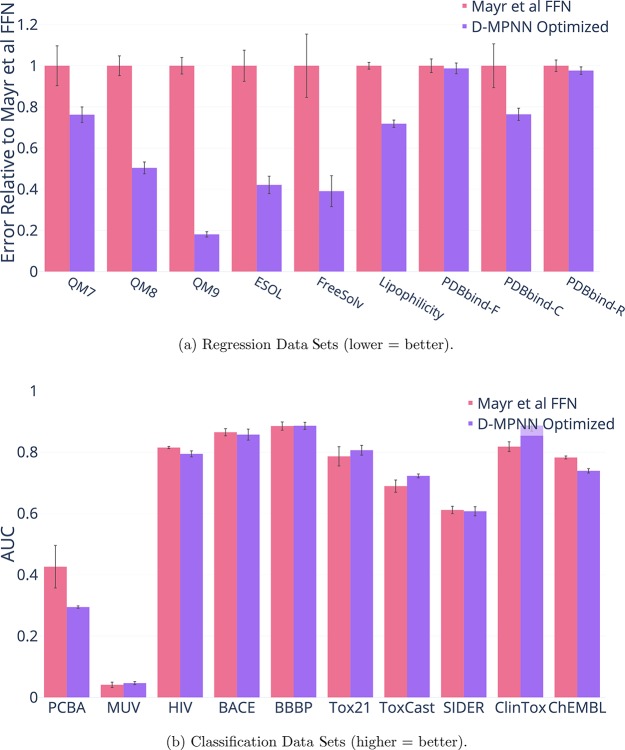

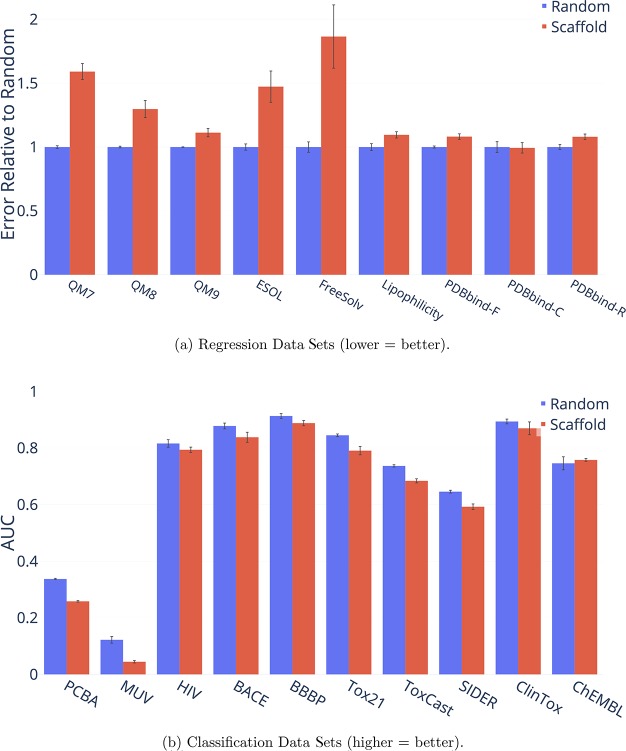
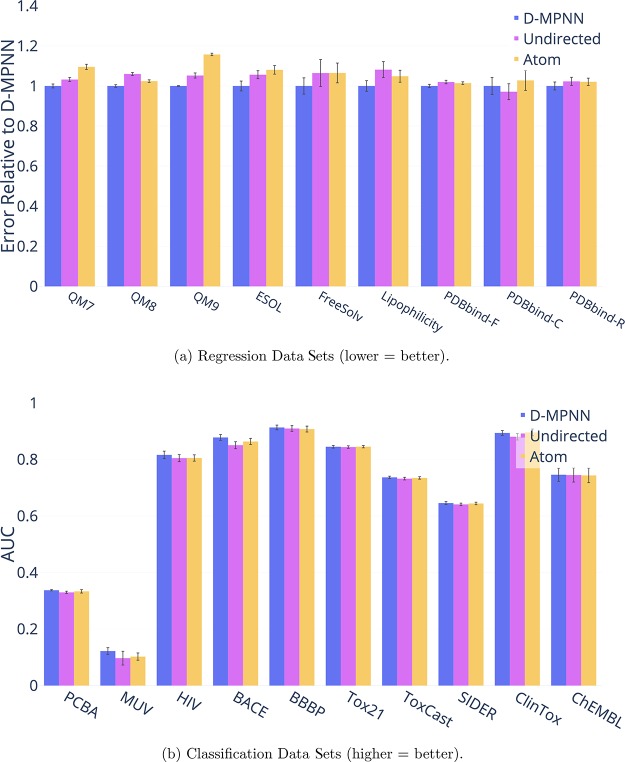
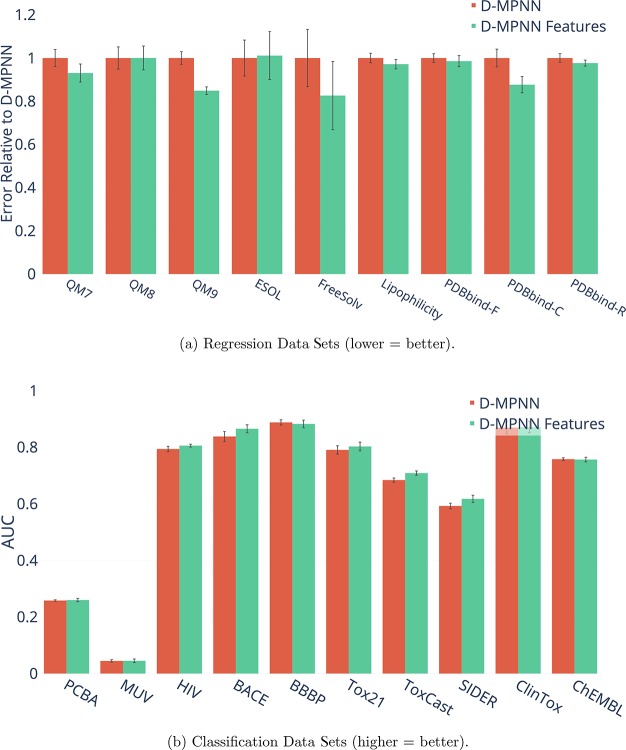
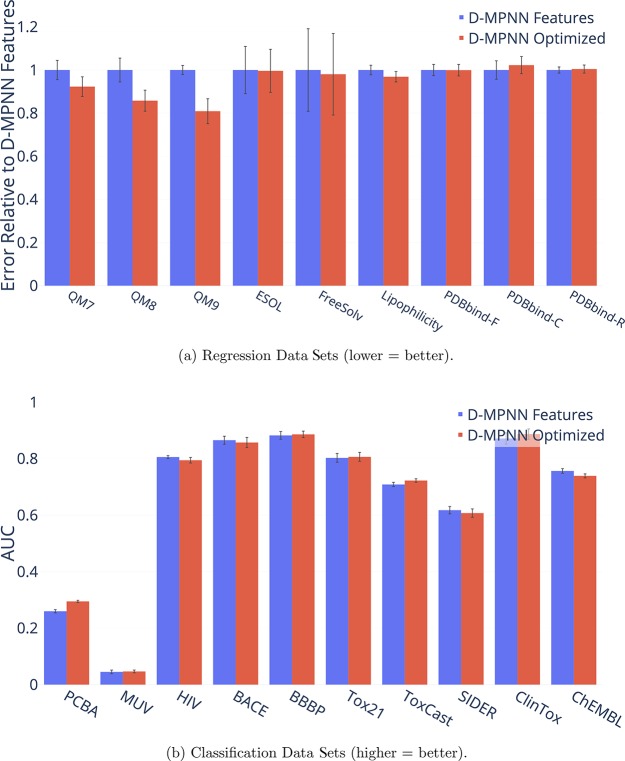
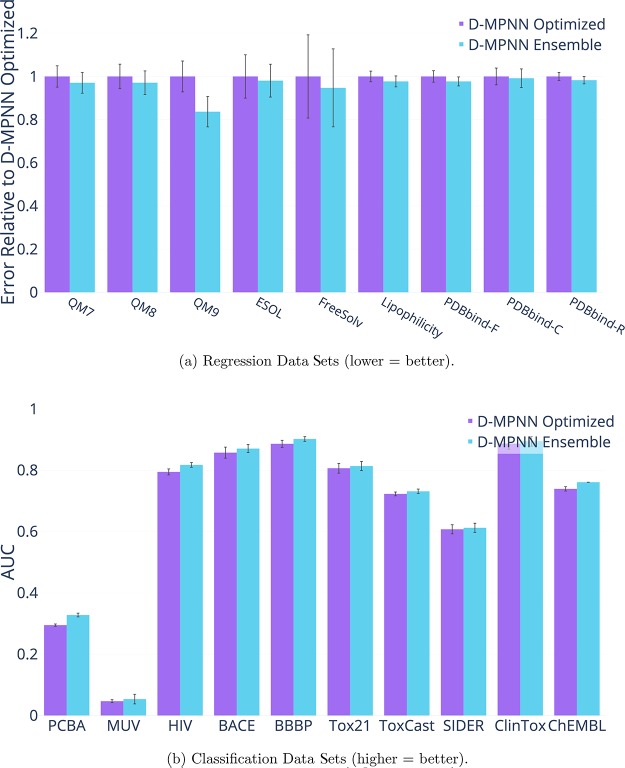
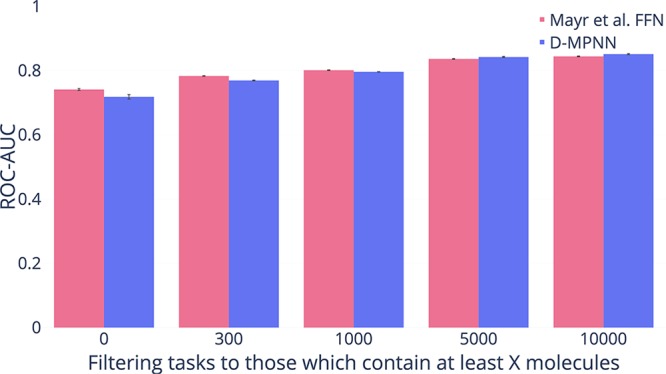
References
-
- Duvenaud D. K.; Maclaurin D.; Iparraguirre J.; Bombarell R.; Hirzel T.; Aspuru-Guzik A.; Adams R. P. Convolutional Networks on Graphs for Learning Molecular Fingerprints. Advances in Neural Information Processing Systems 2015, 2224–2232.
-
- Gilmer J.; Schoenholz S. S.; Riley P. F.; Vinyals O.; Dahl G. E. Neural Message Passing for Quantum Chemistry. Proceedings of the 34th International Conference on Machine Learning 2017, 70, 1263–1272.
-
- Li Y.; Tarlow D.; Brockschmidt M.; Zemel R.. Gated Graph Sequence Neural Networks. 2015, arXiv preprint arXiv:1511.05493. https://arxiv.org/abs/1511.05493 (accessed Aug 6, 2019).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
