

Reduced alphabet of prebiotic amino acids optimally encodes the conformational space of diverse extant protein folds
- PMID: 31362700
- PMCID: PMC6668081
- DOI: 10.1186/s12862-019-1464-6
Reduced alphabet of prebiotic amino acids optimally encodes the conformational space of diverse extant protein folds
Abstract
Background: There is wide agreement that only a subset of the twenty standard amino acids existed prebiotically in sufficient concentrations to form functional polypeptides. We ask how this subset, postulated as {A,D,E,G,I,L,P,S,T,V}, could have formed structures stable enough to found metabolic pathways. Inspired by alphabet reduction experiments, we undertook a computational analysis to measure the structural coding behavior of sequences simplified by reduced alphabets. We sought to discern characteristics of the prebiotic set that would endow it with unique properties relevant to structure, stability, and folding.
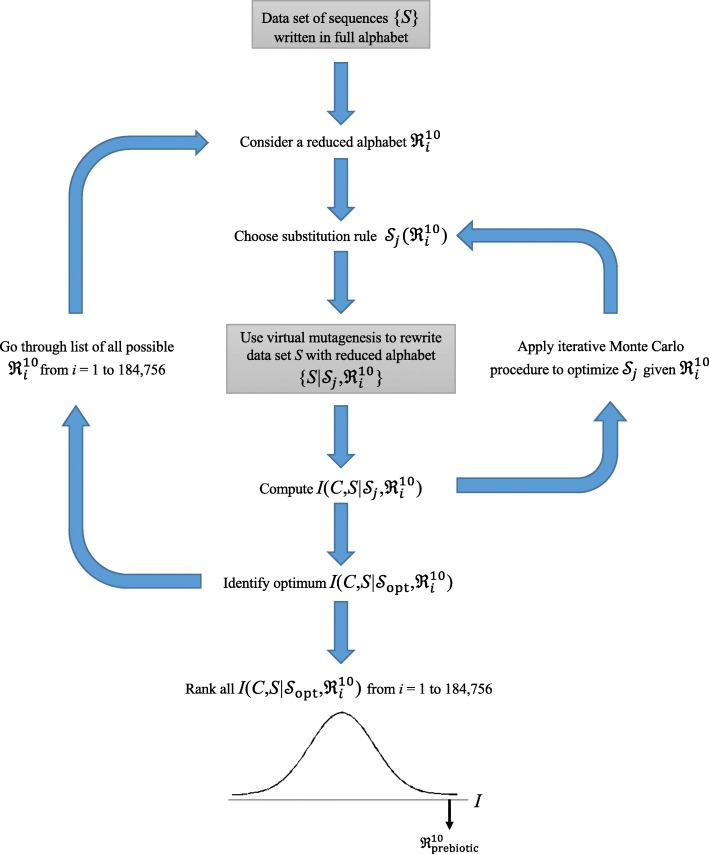
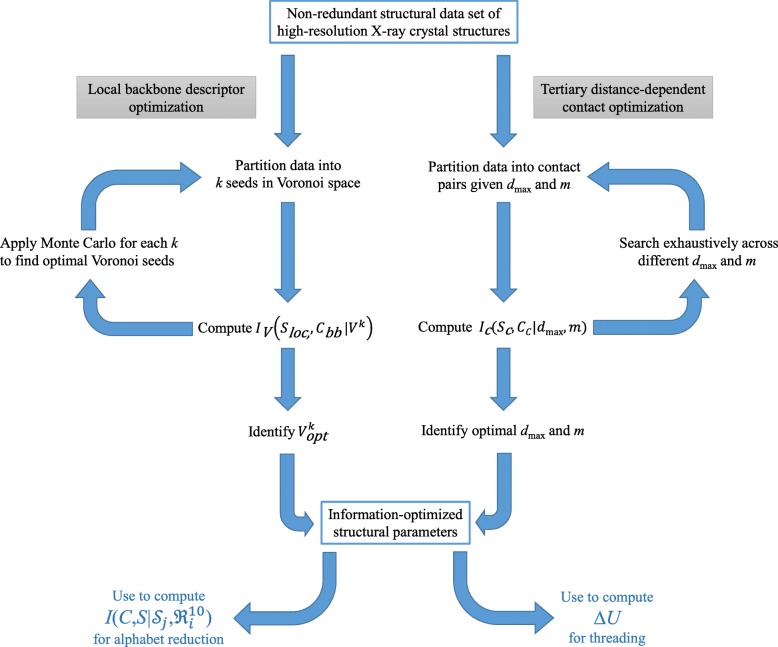
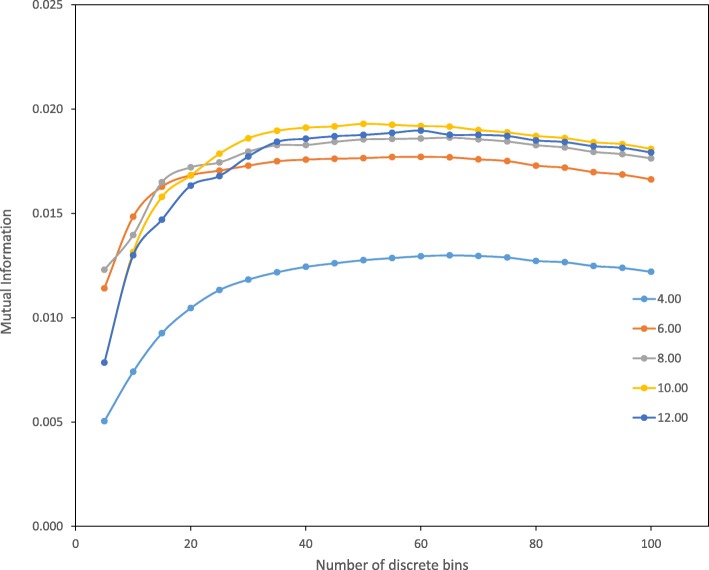
Results: Drawing on a large dataset of single-domain proteins, we employed an information-theoretic measure to assess how well the prebiotic amino acid set preserves fold information against all other possible ten-amino acid sets. An extensive virtual mutagenesis procedure revealed that the prebiotic set excellently preserves sequence-dependent information regarding both backbone conformation and tertiary contact matrix of proteins. We observed that information retention is fold-class dependent: the prebiotic set sufficiently encodes the structure space of α/β and α + β folds, and to a lesser extent, of all-α and all-β folds. The prebiotic set appeared insufficient to encode the small proteins. Assessing how well the prebiotic set discriminates native vs. incorrect sequence-structure matches, we found that α/β and α + β folds exhibit more pronounced energy gaps with the prebiotic set than with nearly all alternatives.
Conclusions: The prebiotic set optimally encodes local backbone structures that appear in the folded environment and near-optimally encodes the tertiary contact matrix of extant proteins. The fold-class-specific patterns observed from our structural analysis confirm the postulated timeline of fold appearance in proteogenesis derived from proteomic sequence analyses. Polypeptides arising in a prebiotic environment will likely form α/β and α + β-like folds if any at all. We infer that the progressive expansion of the alphabet allowed the increased conformational stability and functional specificity of later folds, including all-α, all-β, and small proteins. Our results suggest that prebiotic sequences are amenable to mutations that significantly lower native conformational energies and increase discrimination amidst incorrect folds. This property may have assisted the genesis of functional proto-enzymes prior to the expansion of the full amino acid alphabet.
Keywords: Information theory; Mutual information; Prebiotic amino acids; Protein backbone conformation; Protein evolution; Protein structure; Proteogenesis; Reduced amino acid alphabets; Residue contacts.
Conflict of interest statement
The author declares that he has no competing interests.
Figures

Similar articles
-
Simplified protein design biased for prebiotic amino acids yields a foldable, halophilic protein.Proc Natl Acad Sci U S A. 2013 Feb 5;110(6):2135-9. doi: 10.1073/pnas.1219530110. Epub 2013 Jan 22. Proc Natl Acad Sci U S A. 2013. PMID: 23341608 Free PMC article.
-
Amino acid alphabet reduction preserves fold information contained in contact interactions in proteins.Proteins. 2015 Dec;83(12):2198-216. doi: 10.1002/prot.24936. Proteins. 2015. PMID: 26407535
-
Protein design with L- and D-alpha-amino acid structures as the alphabet.Acc Chem Res. 2008 Oct;41(10):1301-8. doi: 10.1021/ar700265t. Epub 2008 Jul 22. Acc Chem Res. 2008. PMID: 18642934
-
Folding protein alpha-carbon chains into compact forms by Monte Carlo methods.Proteins. 1992 Nov;14(3):409-20. doi: 10.1002/prot.340140310. Proteins. 1992. PMID: 1438179 Review.
-
From local structure to a global framework: recognition of protein folds.J R Soc Interface. 2014 Apr 16;11(95):20131147. doi: 10.1098/rsif.2013.1147. Print 2014 Jun 6. J R Soc Interface. 2014. PMID: 24740960 Free PMC article. Review.
Cited by
-
Protein three-dimensional structures at the origin of life.Interface Focus. 2019 Dec 6;9(6):20190057. doi: 10.1098/rsfs.2019.0057. Epub 2019 Oct 18. Interface Focus. 2019. PMID: 31641431 Free PMC article. Review.
-
Reconstruction and Characterization of Thermally Stable and Catalytically Active Proteins Comprising an Alphabet of ~ 13 Amino Acids.J Mol Evol. 2020 May;88(4):372-381. doi: 10.1007/s00239-020-09938-0. Epub 2020 Mar 23. J Mol Evol. 2020. PMID: 32201904
-
Determination of the Amino Acid Recruitment Order in Early Life by Genome-Wide Analysis of Amino Acid Usage Bias.Biomolecules. 2022 Jan 21;12(2):171. doi: 10.3390/biom12020171. Biomolecules. 2022. PMID: 35204672 Free PMC article.
-
Probing the Role of Cysteine Thiyl Radicals in Biology: Eminently Dangerous, Difficult to Scavenge.Antioxidants (Basel). 2022 Apr 29;11(5):885. doi: 10.3390/antiox11050885. Antioxidants (Basel). 2022. PMID: 35624747 Free PMC article. Review.
-
Early Selection of the Amino Acid Alphabet Was Adaptively Shaped by Biophysical Constraints of Foldability.J Am Chem Soc. 2023 Mar 8;145(9):5320-5329. doi: 10.1021/jacs.2c12987. Epub 2023 Feb 24. J Am Chem Soc. 2023. PMID: 36826345 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
