

Genome-Wide Association and Gene Co-expression Network Analyses Reveal Complex Genetics of Resistance to Goss's Wilt of Maize
- PMID: 31362973
- PMCID: PMC6778796
- DOI: 10.1534/g3.119.400347
Genome-Wide Association and Gene Co-expression Network Analyses Reveal Complex Genetics of Resistance to Goss's Wilt of Maize
Abstract
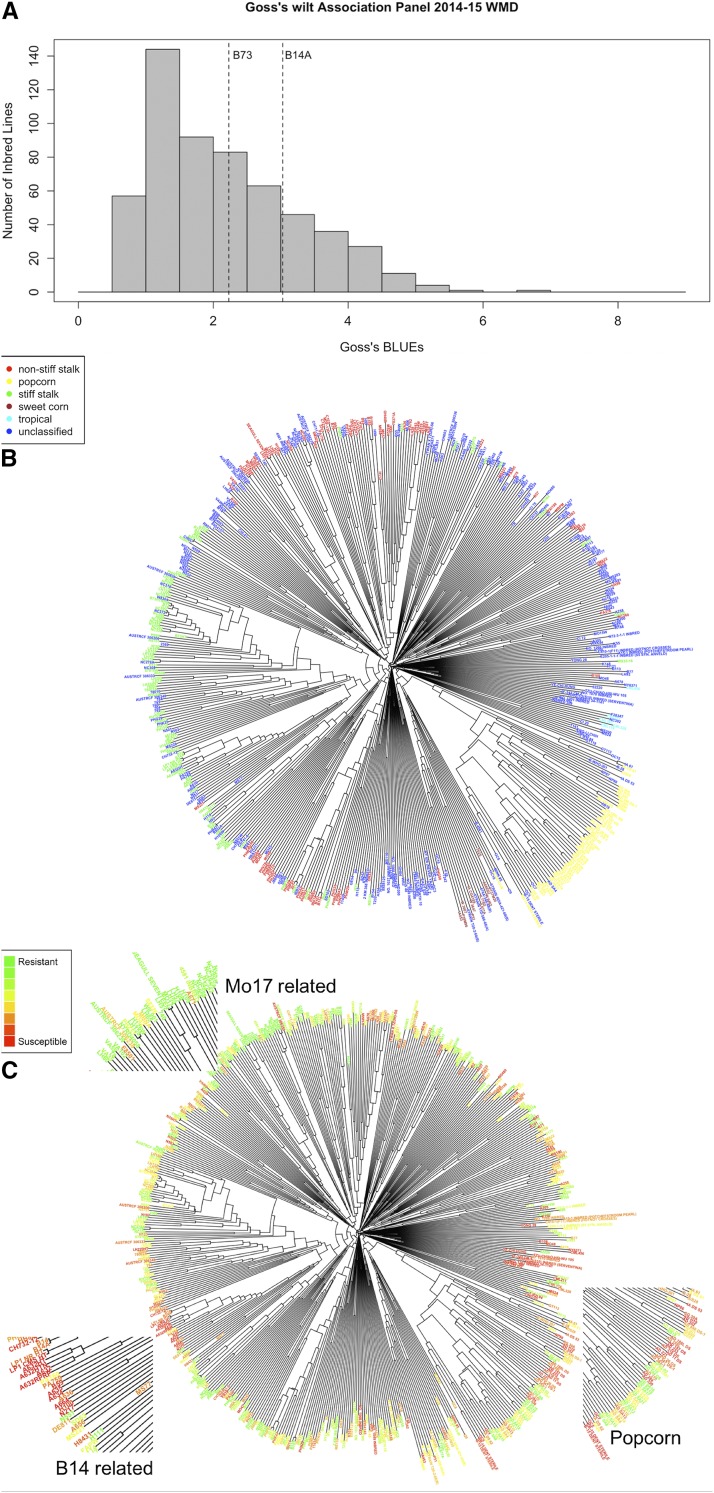
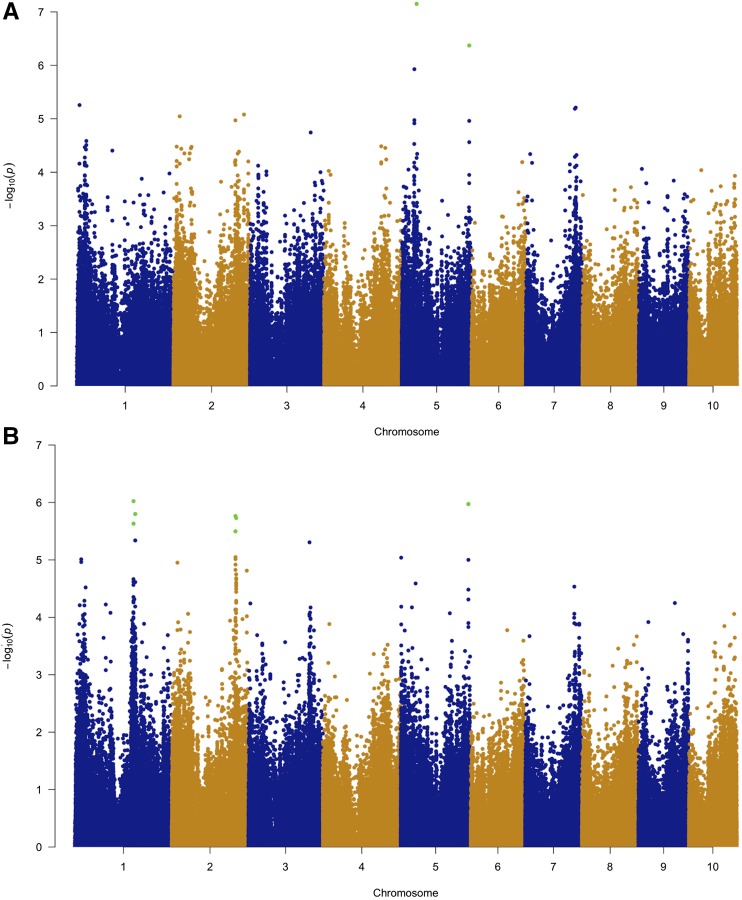
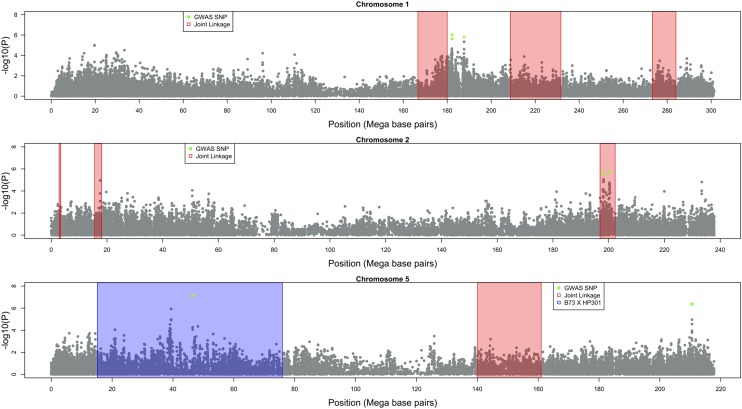
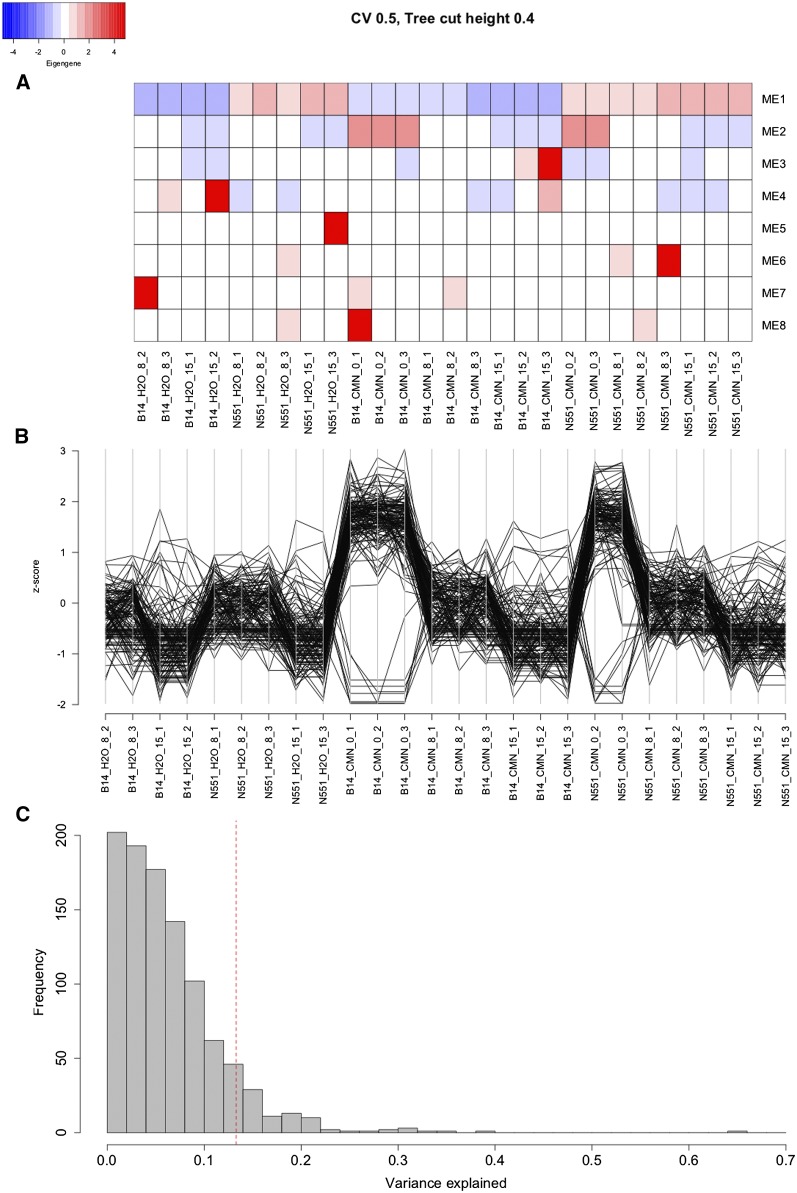
Goss's bacterial wilt and leaf blight is a disease of maize caused by the gram positive bacterium Clavibacter michiganensis subsp. nebraskensis (Cmn). First discovered in Nebraska, Goss's wilt has now spread to major maize growing states in the United States and three provinces in Canada. Previous studies conducted using elite maize inbred lines and their hybrids have shown that resistance to Goss's wilt is a quantitative trait. The objective of this study was to further our understanding of the genetic basis of resistance to Goss's wilt by using a combined approach of genome-wide association mapping and gene co-expression network analysis. Genome-wide association analysis was accomplished using a diversity panel consisting of 555 maize inbred lines and a set of 450 recombinant inbred lines (RILs) from three bi-parental mapping populations, providing the most comprehensive screening of Goss's wilt resistance to date. Three SNPs in the diversity panel and 10 SNPs in the combined dataset, including the diversity panel and RILs, were found to be significantly associated with Goss's wilt resistance. Each significant SNP explained 1-5% of the phenotypic variation for Goss's wilt (total of 8-11%). To augment the results of genome-wide association mapping and help identify candidate genes, a time course RNA sequencing experiment was conducted using resistant (N551) and susceptible (B14A) maize inbred lines. Gene co-expression network analysis of this time course experiment identified one module of 141 correlated genes that showed differential regulation in response to Cmn inoculations in both resistant and susceptible lines. SNPs inside and flanking these genes explained 13.3% of the phenotypic variation. Among 1,000 random samples of genes, only 8% of samples explained more phenotypic variance for Goss's wilt resistance than those implicated by the co-expression network analysis. While a statistically significant enrichment was not observed (P < 0.05), these results suggest a possible role for these genes in quantitative resistance at the field level and warrant more research on combining gene co-expression network analysis with quantitative genetic analyses to dissect complex disease resistance traits. The results of the GWAS and co-expression analysis both support the complex nature of resistance to this important disease of maize.
Keywords: Clavibacter; Goss’s wilt; Maize (Zea mays); genome-wide association mapping; genotyping-by-sequencing; quantitative disease resistance; quantitative trait loci; single nucleotide polymorphism; weighted gene co-expression network analysis.
Copyright © 2019 Singh et al.
Figures
References
-
- Abendroth L. J., Elmore R. W., Boyer M. J., and Marlay S. K., 2011. Corn Growth and Development Iowa State University Press, Ames, https://store.extension.iastate.edu/product/6065.
-
- Balint-Kurti P. J., Yang J., Esbroeck G. V., Jung J., and Smith M. E., 2010. Use of a Maize Advanced Intercross Line for Mapping of QTL for Northern Leaf Blight Resistance and Multiple Disease Resistance. Crop Sci. 50: 458–466. 10.2135/cropsci2009.02.0066 - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources