

p110δ PI3-Kinase Inhibition Perturbs APP and TNFα Trafficking, Reduces Plaque Burden, Dampens Neuroinflammation, and Prevents Cognitive Decline in an Alzheimer's Disease Mouse Model
- PMID: 31363064
- PMCID: PMC6774409
- DOI: 10.1523/JNEUROSCI.0674-19.2019
p110δ PI3-Kinase Inhibition Perturbs APP and TNFα Trafficking, Reduces Plaque Burden, Dampens Neuroinflammation, and Prevents Cognitive Decline in an Alzheimer's Disease Mouse Model
Abstract
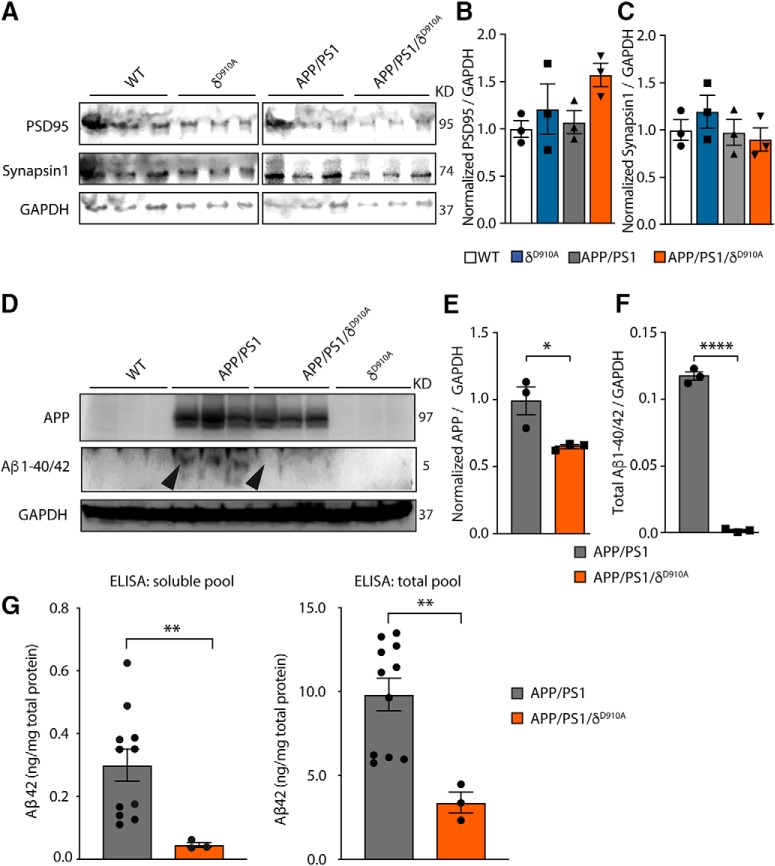
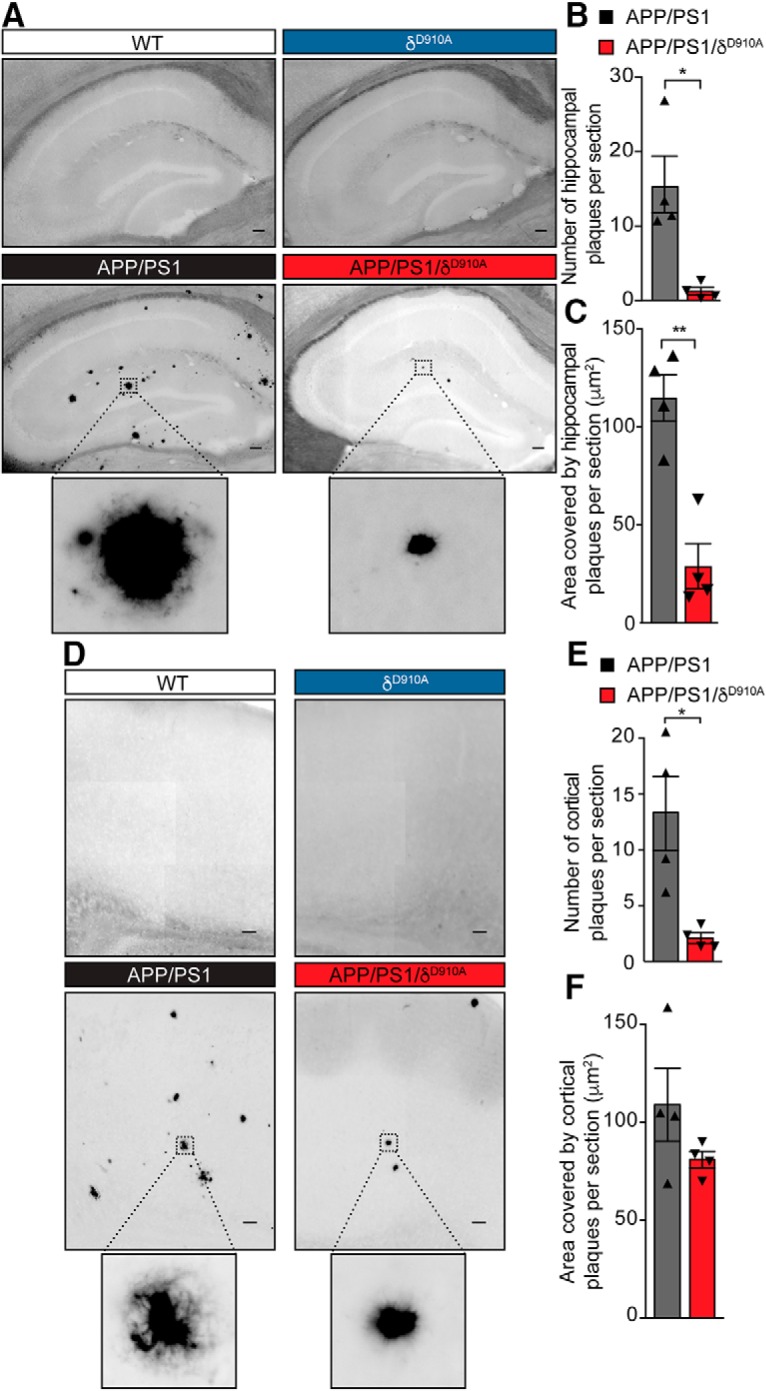
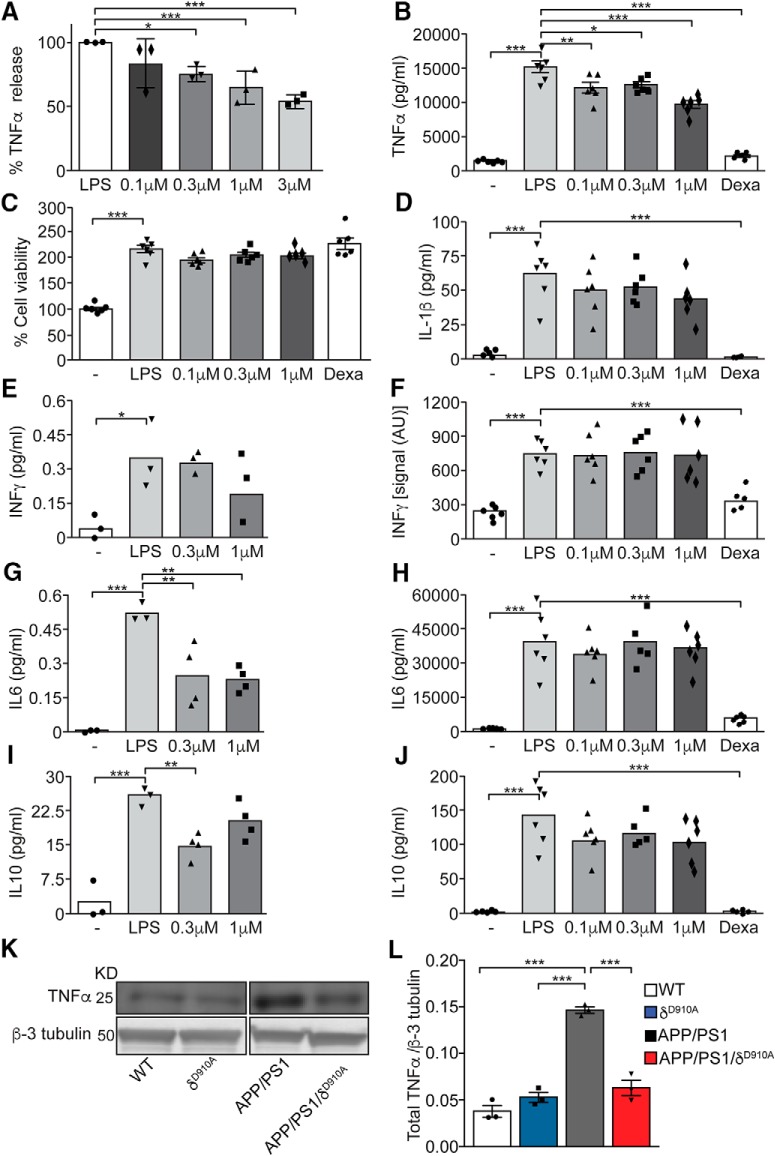
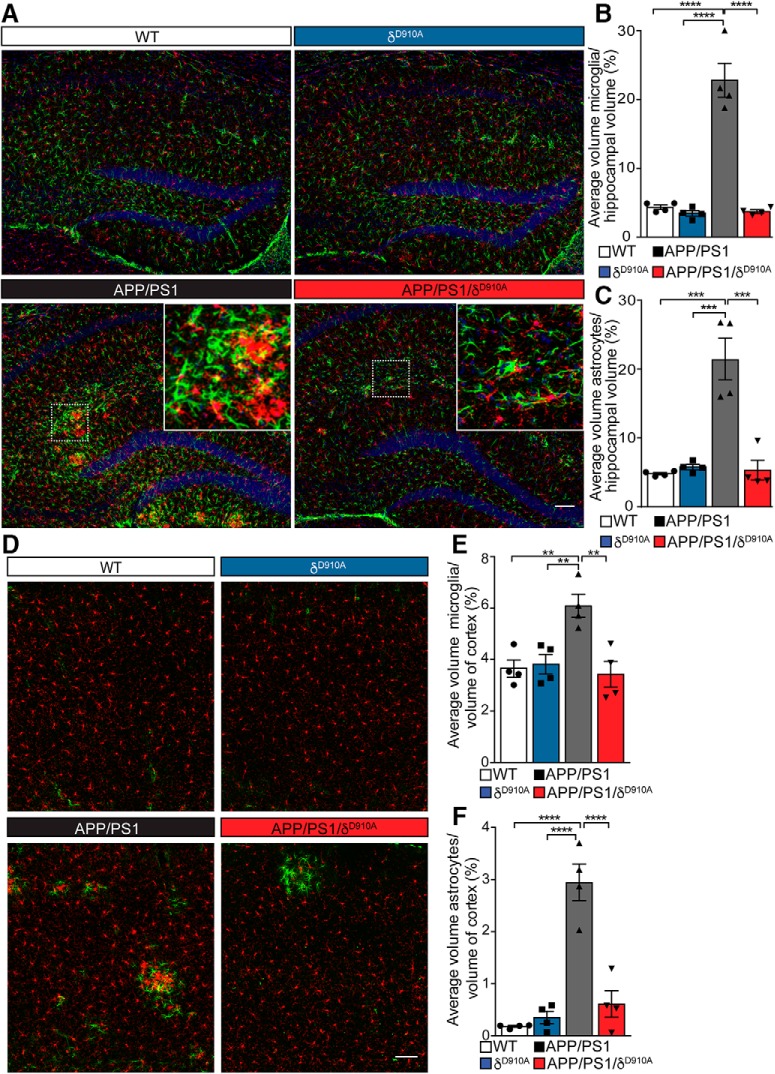
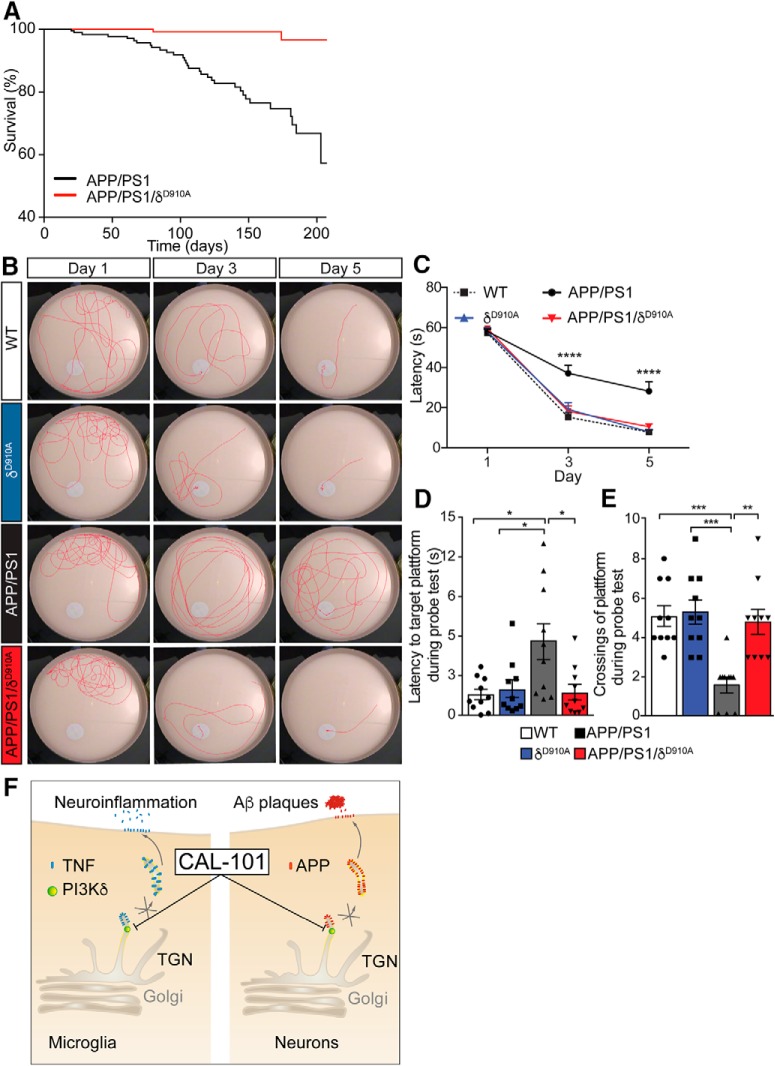
Alzheimer's disease (AD) is associated with the cleavage of the amyloid precursor protein (APP) to produce the toxic amyloid-β (Aβ) peptide. Accumulation of Aβ, together with the concomitant inflammatory response, ultimately leads to neuronal death and cognitive decline. Despite AD progression being underpinned by both neuronal and immunological components, therapeutic strategies based on dual targeting of these systems remains unexplored. Here, we report that inactivation of the p110δ isoform of phosphoinositide 3-kinase (PI3K) reduces anterograde axonal trafficking of APP in hippocampal neurons and dampens secretion of the inflammatory cytokine tumor necrosis factor-alpha by microglial cells in the familial AD APPswe/PS1ΔE9 (APP/PS1) mouse model. Moreover, APP/PS1 mice with kinase-inactive PI3Kδ (δD910A) had reduced Aβ peptides levels and plaques in the brain and an abrogated inflammatory response compared with APP/PS1 littermates. Mechanistic investigations reveal that PI3Kδ inhibition decreases the axonal transport of APP by eliciting the formation of highly elongated tubular-shaped APP-containing carriers, reducing the levels of secreted Aβ peptide. Importantly, APP/PS1/δD910A mice exhibited no spatial learning or memory deficits. Our data highlight inhibition of PI3Kδ as a new approach to protect against AD pathology due to its dual action of dampening microglial-dependent neuroinflammation and reducing plaque burden by inhibition of neuronal APP trafficking and processing.SIGNIFICANCE STATEMENT During Alzheimer's disease (AD), the accumulation of the toxic amyloid-β (Aβ) peptide in plaques is associated with a chronic excessive inflammatory response. Uncovering new drug targets that simultaneously reduce both Aβ plaque load and neuroinflammation holds therapeutic promise. Using a combination of genetic and pharmacological approaches, we found that the p110δ isoform of phosphoinositide 3-kinase (PI3K) is involved in anterograde trafficking of the amyloid precursor protein in neurons and in the secretion of tumor necrosis factor-alpha from microglial cells. Genetic inactivation of PI3Kδ reduces Aβ plaque deposition and abrogates the inflammatory response, resulting in a complete rescue of the life span and spatial memory performance. We conclude that inhibiting PI3Kδ represents a novel therapeutic approach to ameliorate AD pathology by dampening plaque accumulation and microglial-dependent neuroinflammation.
Keywords: Alzheimer's disease; TNF-alpha; amyloid precursor protein; axon trafficking; neuroinflammation; p110delta.
Copyright © 2019 the authors.
Figures
Similar articles
-
Amyloid-β plaque formation and reactive gliosis are required for induction of cognitive deficits in App knock-in mouse models of Alzheimer's disease.BMC Neurosci. 2019 Mar 20;20(1):13. doi: 10.1186/s12868-019-0496-6. BMC Neurosci. 2019. PMID: 30894120 Free PMC article.
-
Cerebral inoculation of human A53T α-synuclein reduces spatial memory decline and amyloid-β aggregation in APP/PS1 transgenic mice of Alzheimer's disease.Brain Res Bull. 2018 Oct;143:116-122. doi: 10.1016/j.brainresbull.2018.10.003. Epub 2018 Oct 24. Brain Res Bull. 2018. PMID: 30366065
-
Benzo(a)pyrene exposure induced neuronal loss, plaque deposition, and cognitive decline in APP/PS1 mice.J Neuroinflammation. 2020 Aug 31;17(1):258. doi: 10.1186/s12974-020-01925-y. J Neuroinflammation. 2020. PMID: 32867800 Free PMC article.
-
Proteolytic processing of Alzheimer's β-amyloid precursor protein.J Neurochem. 2012 Jan;120 Suppl 1(Suppl 1):9-21. doi: 10.1111/j.1471-4159.2011.07519.x. Epub 2011 Nov 28. J Neurochem. 2012. PMID: 22122372 Free PMC article. Review.
-
Nogo receptor interacts with brain APP and Abeta to reduce pathologic changes in Alzheimer's transgenic mice.Curr Alzheimer Res. 2007 Dec;4(5):568-70. doi: 10.2174/156720507783018235. Curr Alzheimer Res. 2007. PMID: 18220524 Free PMC article. Review.
Cited by
-
Efficacy and safety of the integration of traditional Chinese medicine and western medicine in the treatment of diabetes-associated cognitive decline: a systematic review and meta-analysis.Front Pharmacol. 2023 Nov 22;14:1280736. doi: 10.3389/fphar.2023.1280736. eCollection 2023. Front Pharmacol. 2023. PMID: 38074157 Free PMC article. Review.
-
Neuroprotective effect of tormentic acid against memory impairment and neuro‑inflammation in an Alzheimer's disease mouse model.Mol Med Rep. 2020 Aug;22(2):739-750. doi: 10.3892/mmr.2020.11154. Epub 2020 May 18. Mol Med Rep. 2020. PMID: 32468017 Free PMC article.
-
Microglia-Mediated Neuroinflammation Through Phosphatidylinositol 3-Kinase Signaling Causes Cognitive Dysfunction.Int J Mol Sci. 2025 Jul 25;26(15):7212. doi: 10.3390/ijms26157212. Int J Mol Sci. 2025. PMID: 40806341 Free PMC article. Review.
-
Presynaptic APP levels and synaptic homeostasis are regulated by Akt phosphorylation of huntingtin.Elife. 2020 May 26;9:e56371. doi: 10.7554/eLife.56371. Elife. 2020. PMID: 32452382 Free PMC article.
-
Lipids and Secretory Vesicle Exocytosis.Adv Neurobiol. 2023;33:357-397. doi: 10.1007/978-3-031-34229-5_14. Adv Neurobiol. 2023. PMID: 37615874
References
-
- Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, Bouabe H, Scudamore CL, Hancox T, Maecker H, Friedman L, Turner M, Okkenhaug K, Vanhaesebroeck B (2014) Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 510:407–411. 10.1038/nature13444 - DOI - PMC - PubMed
-
- Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, Bales KR, Cappai R, Masters CL, Gliemann J, Mufson EJ, Hyman BT, Paul SM, Nykjaer A, Willnow TE (2005) Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A 102:13461–13466. 10.1073/pnas.0503689102 - DOI - PMC - PubMed
-
- Audrain M, Haure-Mirande JV, Wang M, Kim SH, Fanutza T, Chakrabarty P, Fraser P, St George-Hyslop PH, Golde TE, Blitzer RD, Schadt EE, Zhang B, Ehrlich ME, Gandy S (2018) Integrative approach to sporadic Alzheimer's disease: deficiency of TYROBP in a tauopathy mouse model reduces C1q and normalizes clinical phenotype while increasing spread and state of phosphorylation of tau. Mol Psychiatry. In press. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases