

A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides
- PMID: 31363125
- PMCID: PMC6667459
- DOI: 10.1038/s41467-019-11337-z
A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides
Abstract
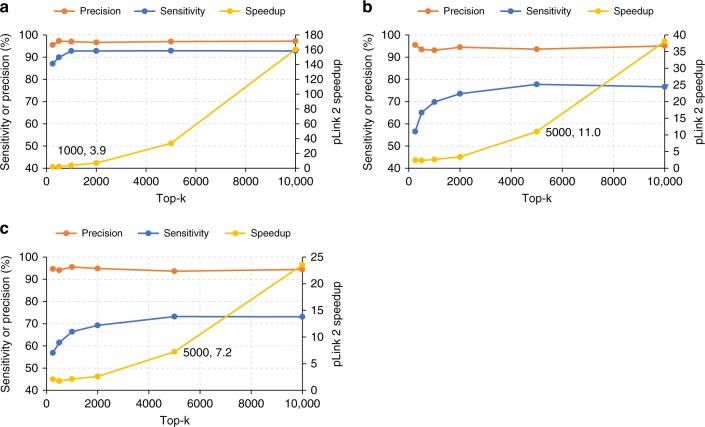
We describe pLink 2, a search engine with higher speed and reliability for proteome-scale identification of cross-linked peptides. With a two-stage open search strategy facilitated by fragment indexing, pLink 2 is ~40 times faster than pLink 1 and 3~10 times faster than Kojak. Furthermore, using simulated datasets, synthetic datasets, 15N metabolically labeled datasets, and entrapment databases, four analysis methods were designed to evaluate the credibility of ten state-of-the-art search engines. This systematic evaluation shows that pLink 2 outperforms these methods in precision and sensitivity, especially at proteome scales. Lastly, re-analysis of four published proteome-scale cross-linking datasets with pLink 2 required only a fraction of the time used by pLink 1, with up to 27% more cross-linked residue pairs identified. pLink 2 is therefore an efficient and reliable tool for cross-linking mass spectrometry analysis, and the systematic evaluation methods described here will be useful for future software development.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
