

Nuclear KIT induces a NFKBIB-RELA-KIT autoregulatory loop in imatinib-resistant gastrointestinal stromal tumors
- PMID: 31363162
- PMCID: PMC6756115
- DOI: 10.1038/s41388-019-0900-9
Nuclear KIT induces a NFKBIB-RELA-KIT autoregulatory loop in imatinib-resistant gastrointestinal stromal tumors
Abstract
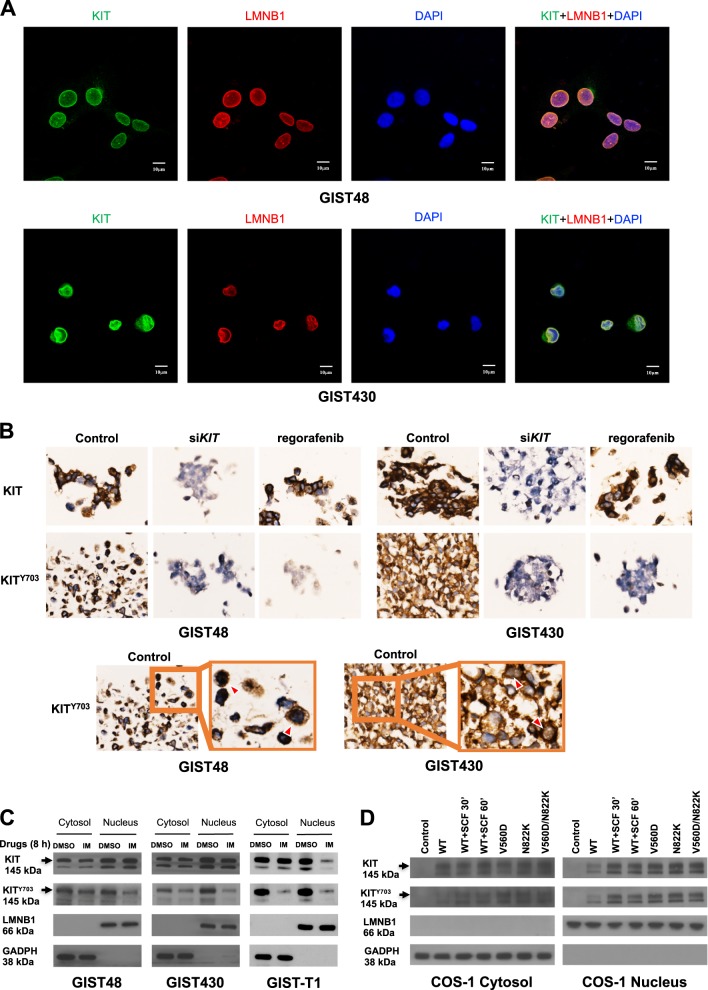
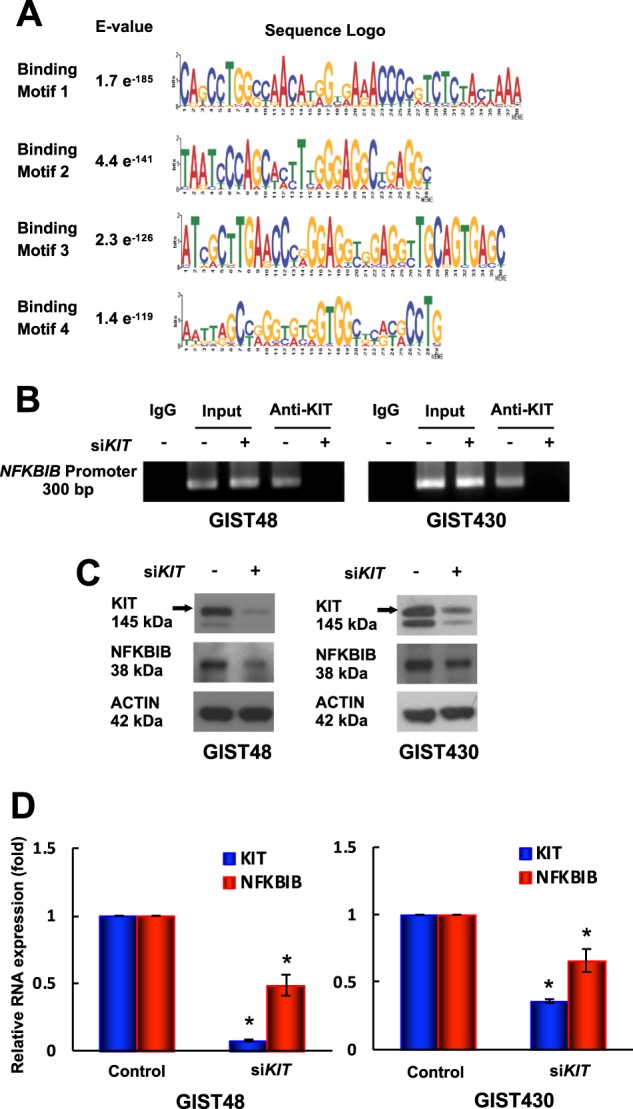
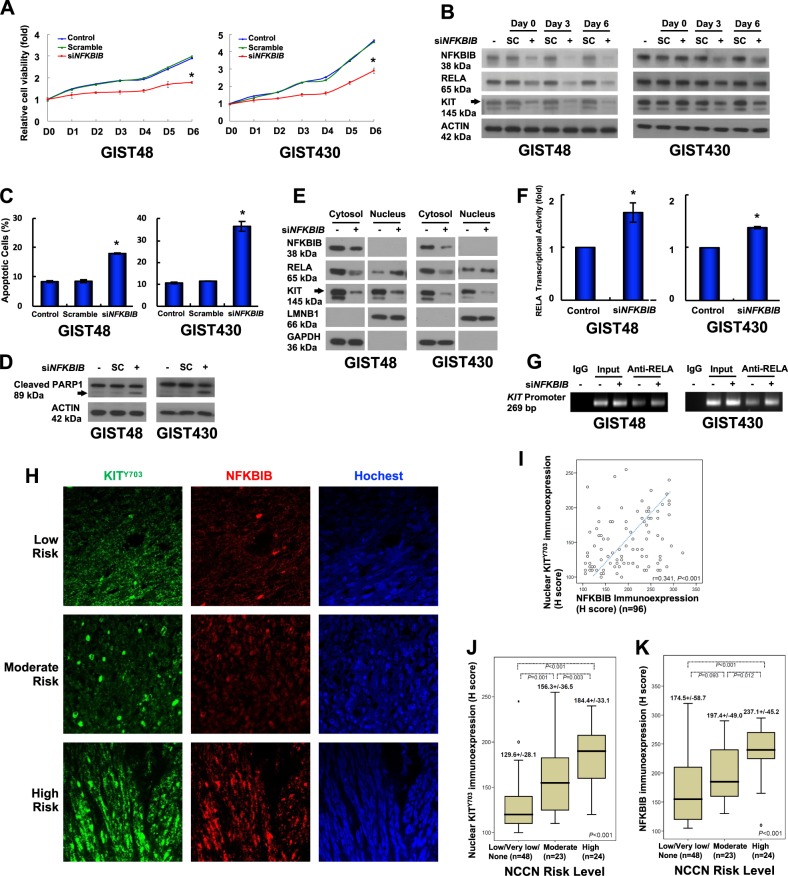
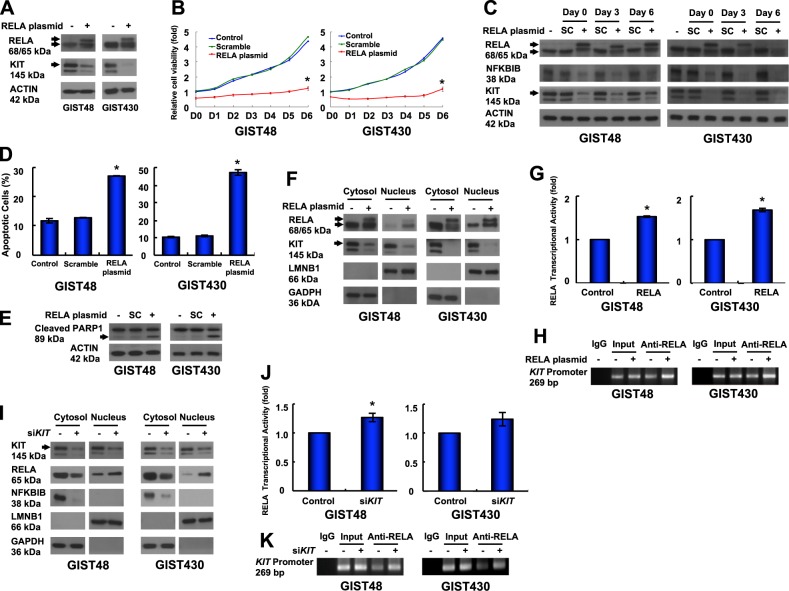
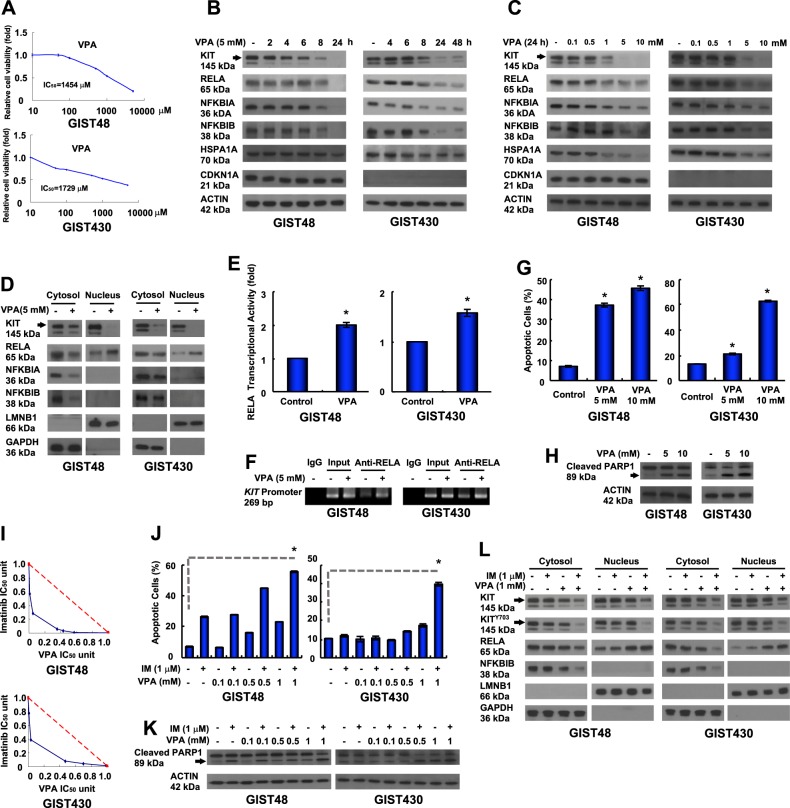
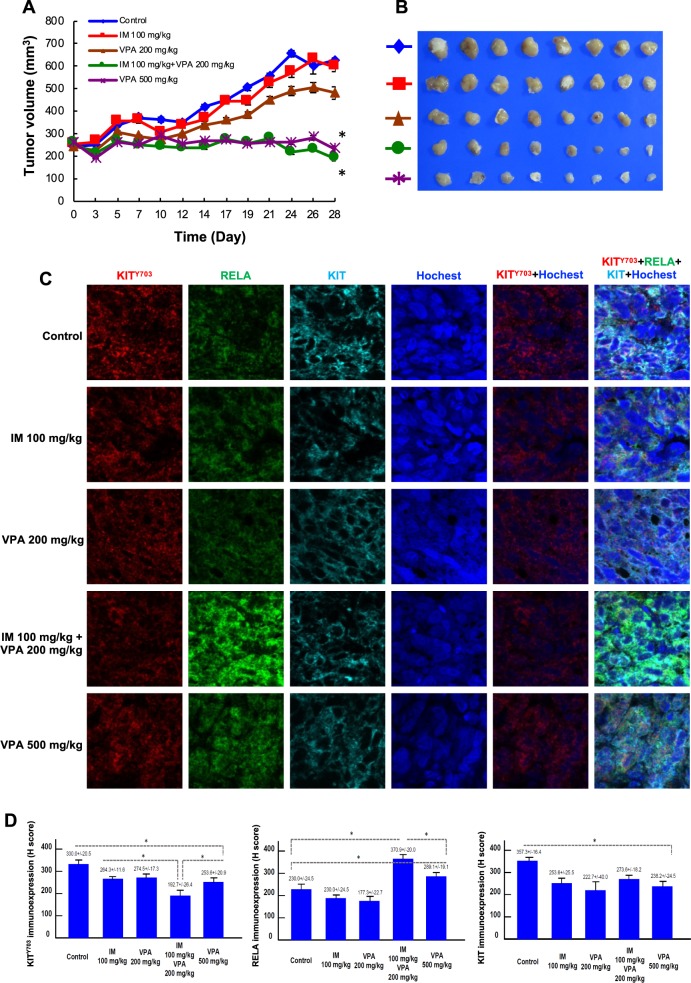
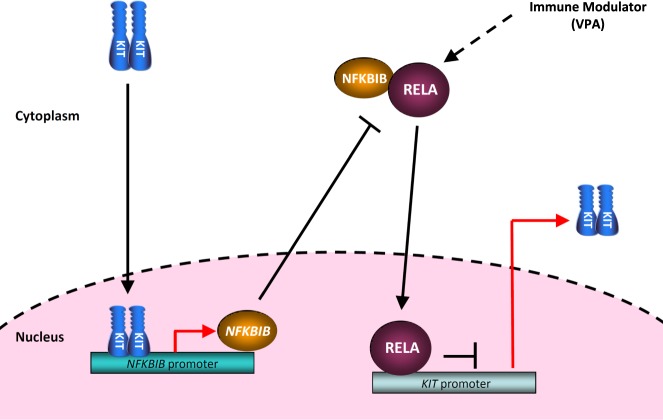
Gastrointestinal stromal tumors (GISTs) are frequently driven by auto-activated, mutant KIT and have durable response to KIT tyrosine kinase inhibitor. However, acquired resistance is an increasing clinical issue in GIST patients receiving front-line imatinib therapy. Our previous studies showed the colocalization of KIT with DAPI-stained nuclei in GIST cells without knowing the role of nuclear KIT in GIST tumorigenesis. In this article, we first identified the binding of nuclear KIT to the promoter of NFKB inhibitor beta (NFKBIB) by chromatin immunoprecipitation (ChIP) sequencing and ChIP assays, which was accompanied with enhanced NFKBIB protein expression in GIST cells. Clinically, high NCCN risk GISTs had significantly higher mean expression levels of nuclear phospho-KIT and NFKBIB as compared with those of intermediate or low/very low-risk GISTs. Conversely, downregulation of NFKBIB by siRNA led to RELA nuclear translocation that could bind to the KIT promoter region and subsequently reduced KIT transcription/expression and the viability of GIST cells. These findings were further confirmed by either RELA overexpression or NFKB/RELA inducer, valproic acid, treatment to result in reduced KIT expression and relative cell viability of imatinib-resistant GIST cells. Combining valproic acid with imatinib showed significantly better growth inhibitory effects on imatinib-resistant GIST48 and GIST430 cells in vitro, and in the GIST430 animal xenograft model. Taken together, these results demonstrate the existence of a nuclear KIT-driven NFKBIB-RELA-KIT autoregulatory loop in GIST tumorigenesis, which are potential targets for developing combination therapy to overcome imatinib-resistant of KIT-expressing GISTs.
Conflict of interest statement
Dr. Li-Tzong Chen reports personal fees from Ono Pharmaceutical, personal fees from Bristol-Myers Squibb, personal fees from Eli Lilly, personal fees from MSD, PharmaEngine, personal fees from Merrimack, grants and personal fees from TTY Biopharm, grants and personal fees from Syncore, personal fees from Five Prime, grants and personal fees from Novartis, personal fees from Pfizer, grants from GlaxoSmithKline, grants from Merck Serono, grants from Polaris, outside the submitted work. The remaining authors declare no potential conflicts of interest.
Figures
Similar articles
-
Dual Targeting of Insulin Receptor and KIT in Imatinib-Resistant Gastrointestinal Stromal Tumors.Cancer Res. 2017 Sep 15;77(18):5107-5117. doi: 10.1158/0008-5472.CAN-17-0917. Epub 2017 Jul 31. Cancer Res. 2017. PMID: 28760855
-
KIT over-expression by p55PIK-PI3K leads to Imatinib-resistance in patients with gastrointestinal stromal tumors.Oncotarget. 2016 Jan 12;7(2):1367-79. doi: 10.18632/oncotarget.6011. Oncotarget. 2016. PMID: 26587973 Free PMC article.
-
Hedgehog pathway dysregulation contributes to the pathogenesis of human gastrointestinal stromal tumors via GLI-mediated activation of KIT expression.Oncotarget. 2016 Nov 29;7(48):78226-78241. doi: 10.18632/oncotarget.12909. Oncotarget. 2016. PMID: 27793025 Free PMC article.
-
The role of KIT in the management of patients with gastrointestinal stromal tumors.Hum Pathol. 2007 May;38(5):679-87. doi: 10.1016/j.humpath.2007.03.001. Hum Pathol. 2007. PMID: 17437861 Review.
-
Emerging Agents for the Treatment of Advanced, Imatinib-Resistant Gastrointestinal Stromal Tumors: Current Status and Future Directions.Drugs. 2015 Aug;75(12):1323-34. doi: 10.1007/s40265-015-0440-8. Drugs. 2015. PMID: 26187774 Free PMC article. Review.
Cited by
-
AXL Inactivation Inhibits Mesothelioma Growth and Migration via Regulation of p53 Expression.Cancers (Basel). 2020 Sep 25;12(10):2757. doi: 10.3390/cancers12102757. Cancers (Basel). 2020. PMID: 32992696 Free PMC article.
-
Fetal hypoxia results in sex- and cell type-specific alterations in neonatal transcription in rat oligodendrocyte precursor cells, microglia, neurons, and oligodendrocytes.Cell Biosci. 2023 Mar 17;13(1):58. doi: 10.1186/s13578-023-01012-8. Cell Biosci. 2023. PMID: 36932456 Free PMC article.
-
Stem cell factor is implicated in microenvironmental interactions and cellular dynamics of chronic lymphocytic leukemia.Haematologica. 2021 Mar 1;106(3):692-700. doi: 10.3324/haematol.2019.236513. Haematologica. 2021. PMID: 32336682 Free PMC article.
-
Overview of current targeted therapy in gallbladder cancer.Signal Transduct Target Ther. 2020 Oct 7;5(1):230. doi: 10.1038/s41392-020-00324-2. Signal Transduct Target Ther. 2020. PMID: 33028805 Free PMC article. Review.
-
Meteorin-like protein overexpression ameliorates fulminant hepatitis in mice by inhibiting chemokine-dependent immune cell infiltration.Acta Pharmacol Sin. 2023 Jul;44(7):1404-1415. doi: 10.1038/s41401-022-01049-4. Epub 2023 Jan 31. Acta Pharmacol Sin. 2023. PMID: 36721008 Free PMC article.
References
-
- Nishida T, Hirota S. Biological and clinical review of stromal tumors in the gastrointestinal tract. Histol Histopathol. 2000;15:1293–301. - PubMed
-
- Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–5. doi: 10.1200/JCO.2007.13.4403. - DOI - PubMed
-
- Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295–302. doi: 10.1016/S0140-6736(12)61857-1. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
