

Gaucher disease in Montenegro - genotype/phenotype correlations: Five cases report
- PMID: 31363476
- PMCID: PMC6656677
- DOI: 10.12998/wjcc.v7.i12.1475
Gaucher disease in Montenegro - genotype/phenotype correlations: Five cases report
Abstract
Background: The most common lysosomal storage disorder is Gaucher disease (GD). It is a deficiency of lysosomal glucocerebrosidase (GBA) due to biallelic mutations in the GBA gene, characterized by the deposition of glucocerebroside in macrophage-monocyte system cells. The report targets clinical phenotypes of GD in order to correlate them with GBA gene mutations, as well as to identify GBA gene mutation in patients in Montenegro that are diagnosed with GD.
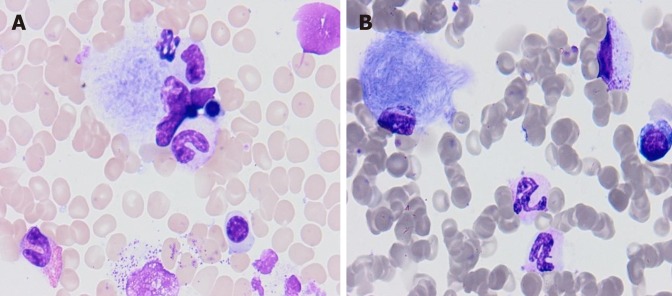
Cases summary: Five patients (4 male, 1 female) of type 1 GD (GD1) are reported. The age at diagnosis ranged from 7 to 40. Patients experienced delays of 1-12 years in diagnosis after the original onset of symptoms. The most common mode of presentation was a variable degree of splenomegaly and thrombocytopenia, while other symptoms included bone pain, hepatomegaly, abdominal pain and fatigue. Osteopenia was present in a majority of the patients: 4/5. All patients were found to have an asymptomatic Erlenmeyer flask deformity of the distal femur. On enzyme replacement therapy (ERT), the hematological and visceral parameters showed significant improvement, but no significant progression in bone mineral density was noticed. GBA gene sequencing revealed homozygosity for the N370S mutation in one patient. The genotypes of the other patients were N370S/55bp deletion, N370S/D409H (2 patients), and H255Q/N370S (1 patient).
Conclusion: The phenotypes of the GD1 encountered in Montenegro were severe but all responded well to ERT.
Keywords: Case report; GBA gene sequencing; Gaucher disease; Genotype; Glucocerebrosidase; Lysosomal storage disorder.
Conflict of interest statement
Conflict-of-interest statement: The authors have no conflicts of interest to declare.
Figures
Similar articles
-
Gaucher disease: Biochemical and molecular findings in 141 patients diagnosed in Greece.Mol Genet Metab Rep. 2020 Jun 7;24:100614. doi: 10.1016/j.ymgmr.2020.100614. eCollection 2020 Sep. Mol Genet Metab Rep. 2020. PMID: 32547927 Free PMC article.
-
Gaucher disease in North Macedonia: Unexpected prevalence of the N370S GBA1 allele with attenuated disease expression.Mol Genet Metab Rep. 2022 Jul 8;32:100895. doi: 10.1016/j.ymgmr.2022.100895. eCollection 2022 Sep. Mol Genet Metab Rep. 2022. PMID: 35845720 Free PMC article.
-
Genotypes and phenotypes in 20 Chinese patients with type 2 Gaucher disease.Brain Dev. 2018 Nov;40(10):876-883. doi: 10.1016/j.braindev.2018.06.006. Epub 2018 Jun 19. Brain Dev. 2018. PMID: 29934114
-
Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA).Hum Mutat. 2008 May;29(5):567-83. doi: 10.1002/humu.20676. Hum Mutat. 2008. PMID: 18338393 Review.
-
Gaucher disease among Chinese patients: review on genotype/phenotype correlation from 29 patients and identification of novel and rare alleles.Blood Cells Mol Dis. 2007 May-Jun;38(3):287-93. doi: 10.1016/j.bcmd.2006.11.003. Epub 2006 Dec 29. Blood Cells Mol Dis. 2007. PMID: 17196853 Review.
Cited by
-
Current opinion on pluripotent stem cell technology in Gaucher's disease: challenges and future prospects.Cytotechnology. 2025 Feb;77(1):26. doi: 10.1007/s10616-024-00687-2. Epub 2024 Dec 27. Cytotechnology. 2025. PMID: 39735330 Review.
-
Development and evaluation of a patient-reported outcome measure specific for Gaucher disease with or without neurological symptoms in Japan.Orphanet J Rare Dis. 2024 Jan 5;19(1):11. doi: 10.1186/s13023-023-02996-9. Orphanet J Rare Dis. 2024. PMID: 38183145 Free PMC article.
-
Screening for Gaucher Disease Using Dried Blood Spot Tests: A Japanese Multicenter, Cross-sectional Survey.Intern Med. 2021;60(5):699-707. doi: 10.2169/internalmedicine.5064-20. Epub 2021 Mar 1. Intern Med. 2021. PMID: 33642560 Free PMC article.
References
-
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. - PubMed
-
- Mistry PK, Liu J, Yang M, Nottoli T, McGrath J, Jain D, Zhang K, Keutzer J, Chuang WL, Mehal WZ, Zhao H, Lin A, Mane S, Liu X, Peng YZ, Li JH, Agrawal M, Zhu LL, Blair HC, Robinson LJ, Iqbal J, Sun L, Zaidi M. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc Natl Acad Sci U S A. 2010;107:19473–19478. - PMC - PubMed
-
- Grabowski GA. Gaucher disease and other storage disorders. Hematology Am Soc Hematol Educ Program. 2012;2012:13–18. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
