

AAV9-Stathmin1 gene delivery improves disease phenotype in an intermediate mouse model of spinal muscular atrophy
- PMID: 31363739
- PMCID: PMC6935388
- DOI: 10.1093/hmg/ddz188
AAV9-Stathmin1 gene delivery improves disease phenotype in an intermediate mouse model of spinal muscular atrophy
Abstract
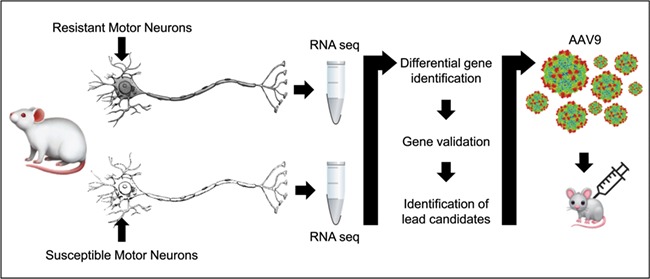
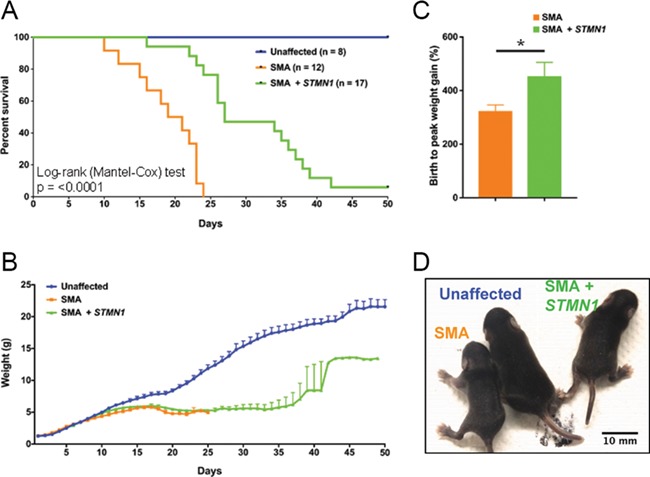
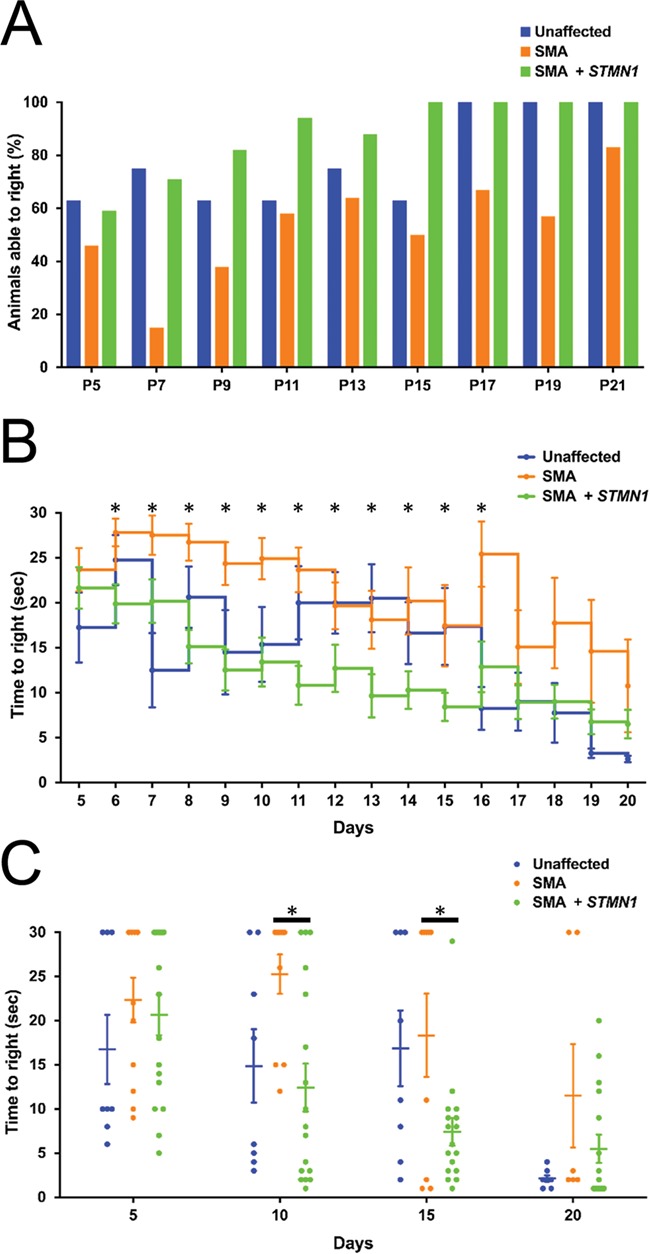
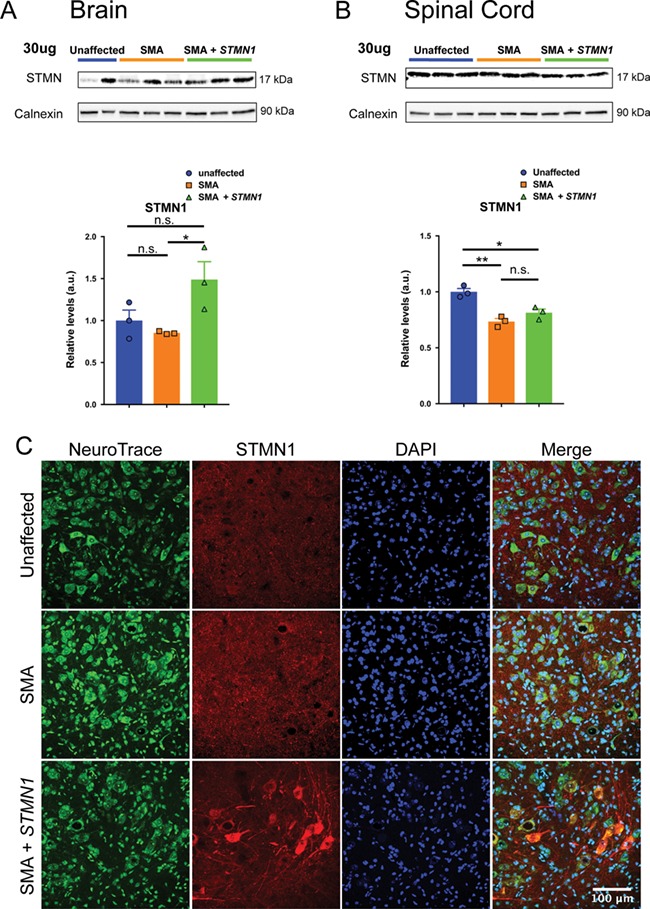
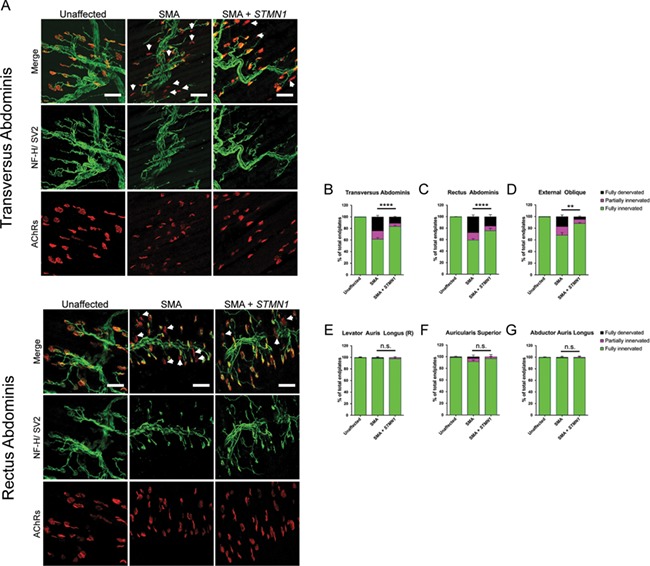
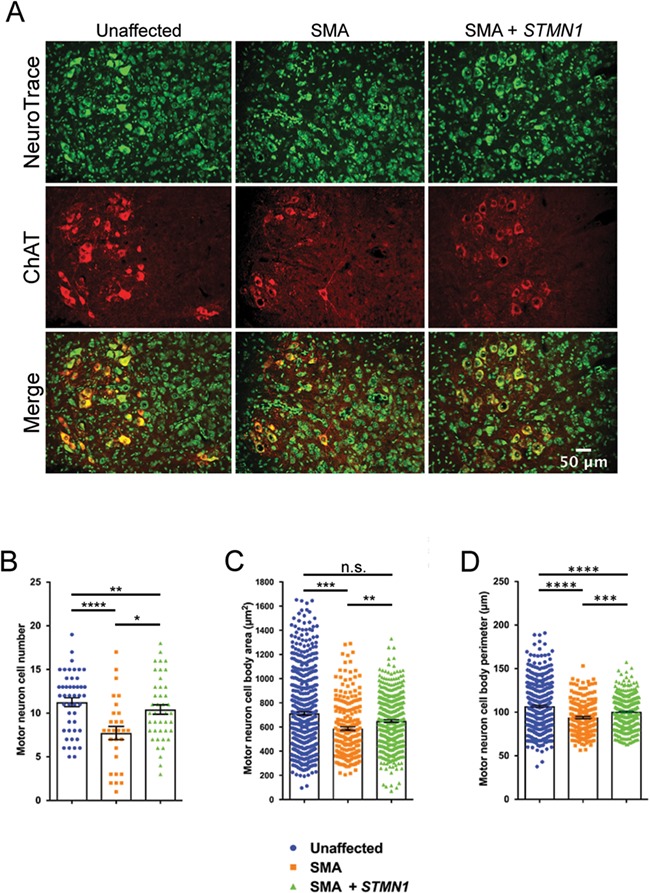
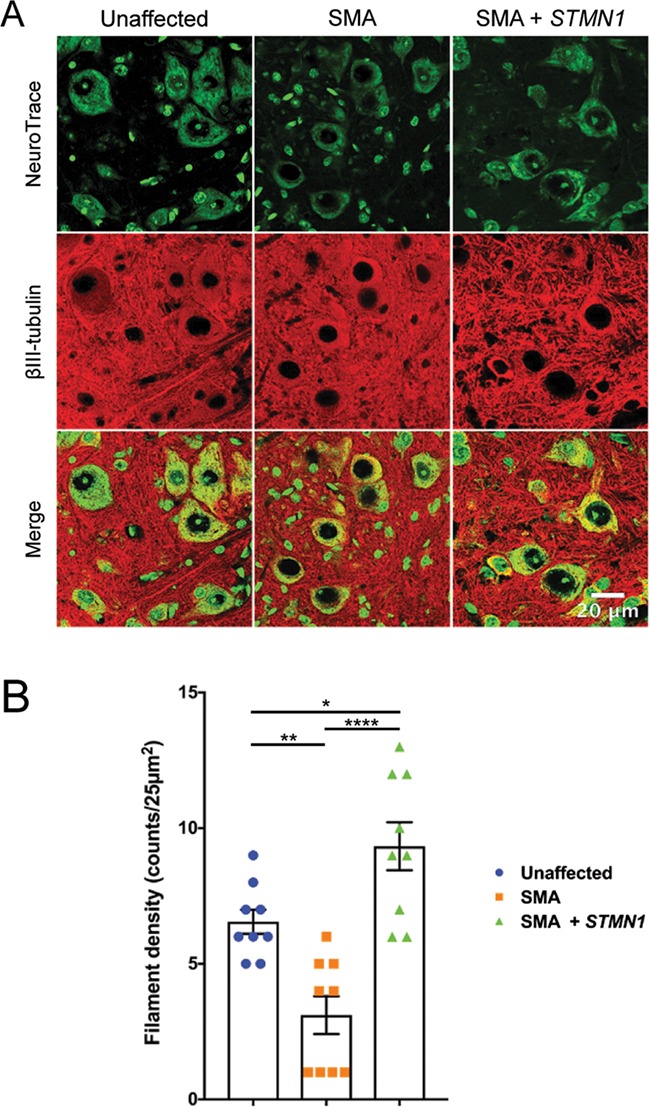
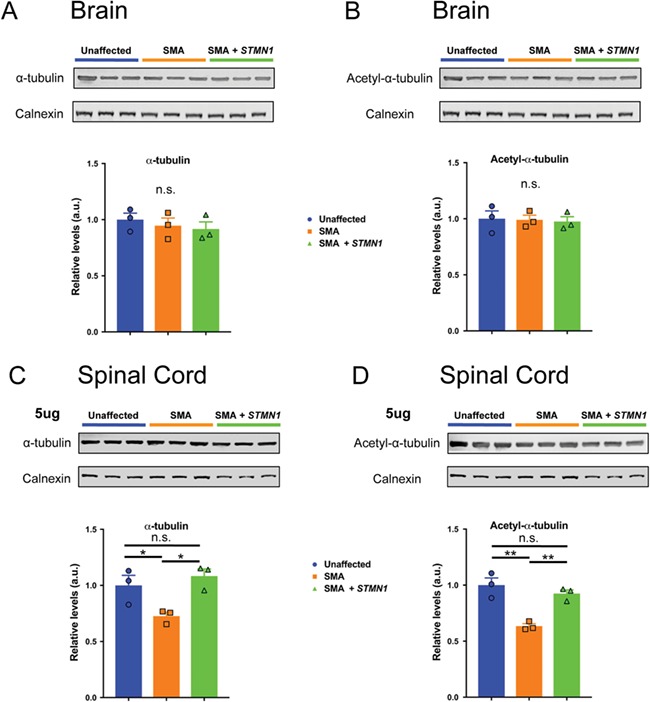
Spinal muscular atrophy (SMA) is a devastating infantile genetic disorder caused by the loss of survival motor neuron (SMN) protein that leads to premature death due to loss of motor neurons and muscle atrophy. The approval of an antisense oligonucleotide therapy for SMA was an important milestone in SMA research; however, effective next-generation therapeutics will likely require combinatorial SMN-dependent therapeutics and SMN-independent disease modifiers. A recent cross-disease transcriptomic analysis identified Stathmin-1 (STMN1), a tubulin-depolymerizing protein, as a potential disease modifier across different motor neuron diseases, including SMA. Here, we investigated whether viral-based delivery of STMN1 decreased disease severity in a well-characterized SMA mouse model. Intracerebroventricular delivery of scAAV9-STMN1 in SMA mice at P2 significantly increased survival and weight gain compared to untreated SMA mice without elevating Smn levels. scAAV9-STMN1 improved important hallmarks of disease, including motor function, NMJ pathology and motor neuron cell preservation. Furthermore, scAAV9-STMN1 treatment restored microtubule networks and tubulin expression without affecting tubulin stability. Our results show that scAAV9-STMN1 treatment improves SMA pathology possibly by increasing microtubule turnover leading to restored levels of stable microtubules. Overall, these data demonstrate that STMN1 can significantly reduce the SMA phenotype independent of restoring SMN protein and highlight the importance of developing SMN-independent therapeutics for the treatment of SMA.
© The Author(s) 2019. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
Cytoskeleton dysfunction of motor neuron in spinal muscular atrophy.J Neurol. 2024 Dec 12;272(1):19. doi: 10.1007/s00415-024-12724-3. J Neurol. 2024. PMID: 39666039 Free PMC article. Review.
-
Decreased stathmin expression ameliorates neuromuscular defects but fails to prolong survival in a mouse model of spinal muscular atrophy.Neurobiol Dis. 2013 Apr;52:94-103. doi: 10.1016/j.nbd.2012.11.015. Epub 2012 Dec 22. Neurobiol Dis. 2013. PMID: 23268200
-
AAV9-Mediated Expression of SMN Restricted to Neurons Does Not Rescue the Spinal Muscular Atrophy Phenotype in Mice.Mol Ther. 2020 Aug 5;28(8):1887-1901. doi: 10.1016/j.ymthe.2020.05.011. Epub 2020 May 15. Mol Ther. 2020. PMID: 32470325 Free PMC article.
-
Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice.Hum Mol Genet. 2011 Feb 15;20(4):681-93. doi: 10.1093/hmg/ddq514. Epub 2010 Nov 30. Hum Mol Genet. 2011. PMID: 21118896
-
Therapy development for spinal muscular atrophy in SMN independent targets.Neural Plast. 2012;2012:456478. doi: 10.1155/2012/456478. Epub 2012 May 31. Neural Plast. 2012. PMID: 22701806 Free PMC article. Review.
Cited by
-
Nucleic Acid-Based Therapeutics in Orphan Neurological Disorders: Recent Developments.Front Mol Biosci. 2021 Apr 28;8:643681. doi: 10.3389/fmolb.2021.643681. eCollection 2021. Front Mol Biosci. 2021. PMID: 33996898 Free PMC article. Review.
-
Mutations in PTPN11 could lead to a congenital myasthenic syndrome phenotype: a Noonan syndrome case series.J Neurol. 2024 Mar;271(3):1331-1341. doi: 10.1007/s00415-023-12070-w. Epub 2023 Nov 4. J Neurol. 2024. PMID: 37923938
-
Cytoskeleton dysfunction of motor neuron in spinal muscular atrophy.J Neurol. 2024 Dec 12;272(1):19. doi: 10.1007/s00415-024-12724-3. J Neurol. 2024. PMID: 39666039 Free PMC article. Review.
-
Mouse models of SMA show divergent patterns of neuronal vulnerability and resilience.Skelet Muscle. 2022 Sep 12;12(1):22. doi: 10.1186/s13395-022-00305-9. Skelet Muscle. 2022. PMID: 36089582 Free PMC article.
-
Stathmins and Motor Neuron Diseases: Pathophysiology and Therapeutic Targets.Biomedicines. 2022 Mar 19;10(3):711. doi: 10.3390/biomedicines10030711. Biomedicines. 2022. PMID: 35327513 Free PMC article. Review.
References
-
- Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. et al. (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell, 80, 155–165. - PubMed
-
- Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H. and McPherson J.D. (1999) A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet., 8, 1177–1183. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
