

Comprehensive genetic analyses using targeted next-generation sequencing and genotype-phenotype correlations in 53 Japanese patients with osteogenesis imperfecta
- PMID: 31363794
- PMCID: PMC7083816
- DOI: 10.1007/s00198-019-05076-6
Comprehensive genetic analyses using targeted next-generation sequencing and genotype-phenotype correlations in 53 Japanese patients with osteogenesis imperfecta
Erratum in
-
Correction to: Comprehensive genetic analyses using targeted next-generation sequencing and genotype-phenotype correlations in 53 Japanese patients with osteogenesis imperfecta.Osteoporos Int. 2020 Jun;31(6):1185. doi: 10.1007/s00198-020-05396-y. Osteoporos Int. 2020. PMID: 32246166 Free PMC article.
Abstract
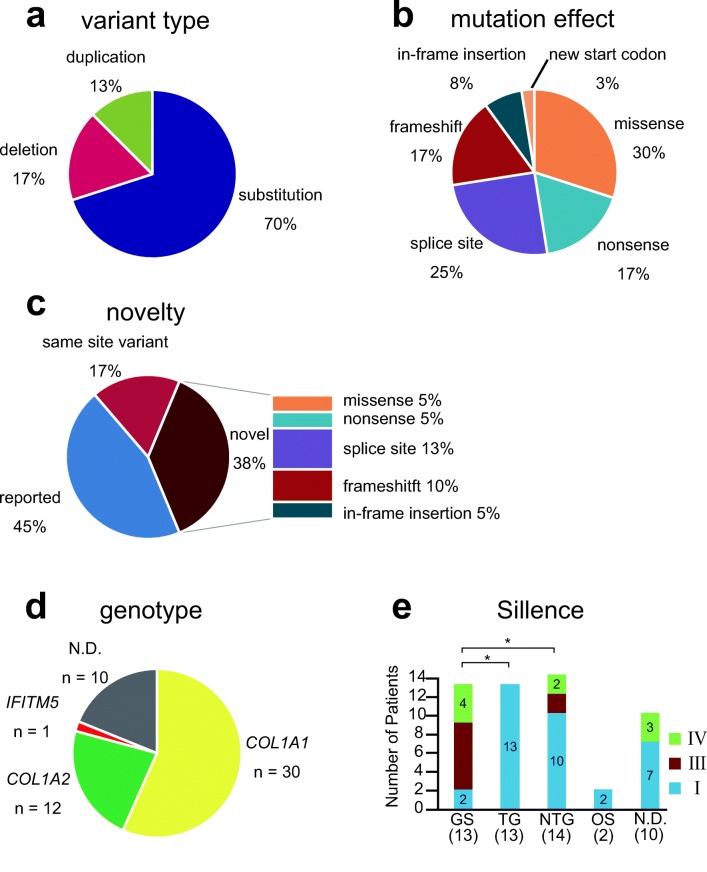
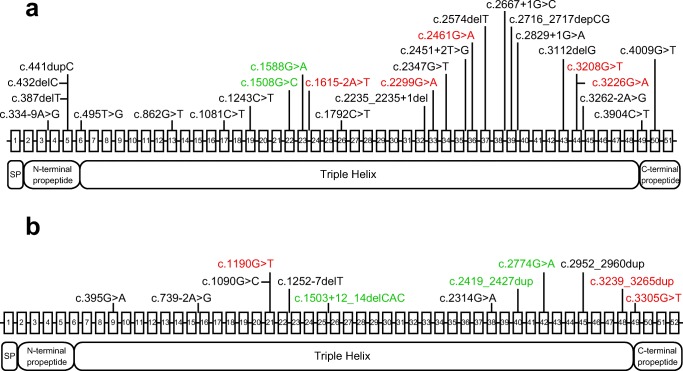
To elucidate mutation spectrum and genotype-phenotype correlations in Japanese patients with OI, we conducted comprehensive genetic analyses using NGS, as this had not been analyzed comprehensively in this patient population. Most mutations were located on COL1A1 and COL1A2. Glycine substitutions in COL1A1 resulted in the severe phenotype.
Introduction: Most cases of osteogenesis imperfecta (OI) are caused by mutations in COL1A1 or COL1A2, which encode α chains of type I collagen. However, mutations in at least 16 other genes also cause OI. The mutation spectrum in Japanese patients with OI has not been comprehensively analyzed, as it is difficult to identify using classical Sanger sequencing. In this study, we aimed to reveal the mutation spectrum and genotype-phenotype correlations in Japanese patients with OI using next-generation sequencing (NGS).
Methods: We designed a capture panel for sequencing 15 candidate OI genes and 19 candidate genes that are associated with bone fragility or Wnt signaling. Using NGS, we examined 53 Japanese patients with OI from unrelated families.
Results: Pathogenic mutations were detected in 43 out of 53 individuals. All mutations were heterozygous. Among the 43 individuals, 40 variants were identified including 15 novel mutations. We found these mutations in COL1A1 (n = 30, 69.8%), COL1A2 (n = 12, 27.9%), and IFITM5 (n = 1, 2.3%). Patients with glycine substitution on COL1A1 had a higher frequency of fractures and were more severely short-statured. Although no significant genotype-phenotype correlation was observed for bone mineral density, the trabecular bone score was significantly lower in patients with glycine substitutions.
Conclusion: We identified pathogenic mutations in 81% of our Japanese patients with OI. Most mutations were located on COL1A1 and COL1A2. This study revealed that glycine substitutions on COL1A1 resulted in the severe phenotype among Japanese patients with OI.
Keywords: Fracture; Genotype-phenotype correlation; Next-generation sequencing; Osteogenesis imperfecta; Short stature; Type I collagen.
Conflict of interest statement
None.
Figures
References
-
- Marini JC (2018) Osteogenesis imperfecta. In: Bilezikian JP (ed) Primer on the metabolic bone diseases and disorders of mineral metabolism. Wiley-Blackwell, pp 871–877
-
- Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, Spranger J, Superti-Furga A, Warman M, Unger S. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167A:2869–2892. doi: 10.1002/ajmg.a.37365. - DOI - PubMed
-
- Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, Hyland JC, Korkko J, Prockop DJ, De Paepe A, Coucke P, Symoens S, Glorieux FH, Roughley PJ, Lund AM, Kuurila-Svahn K, Hartikka H, Cohn DH, Krakow D, Mottes M, Schwarze U, Chen D, Yang K, Kuslich C, Troendle J, Dalgleish R, Byers PH. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28:209–221. doi: 10.1002/humu.20429. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
