

DNA Damage Repair in Huntington's Disease and Other Neurodegenerative Diseases
- PMID: 31364066
- PMCID: PMC6985310
- DOI: 10.1007/s13311-019-00768-7
DNA Damage Repair in Huntington's Disease and Other Neurodegenerative Diseases
Abstract
Recent genome-wide association studies of Huntington's disease (HD) primarily highlighted genes involved in DNA damage repair mechanisms as modifiers of age at onset and disease severity, consistent with evidence that more DNA repair genes are being implicated in late age-onset neurodegenerative diseases. This provides an exciting opportunity to advance therapeutic development in HD, as these pathways have already been under intense investigation in cancer research. Also emerging are the roles of other polyglutamine disease proteins in DNA damage repair mechanisms. A potential universal trigger of oxidative DNA damage shared in these late age-onset diseases is the increase of reactive oxygen species (ROS) in human aging, defining an age-related mechanism that has defied other hypotheses of neurodegeneration. We discuss the potential commonality of DNA damage repair pathways in HD and other neurodegenerative diseases. Potential targets for therapy that may prove beneficial across many of these diseases are also identified, defining nodes in the ataxia telangiectasia-mutated (ATM) complex, mismatch repair, and poly ADP-ribose polymerases (PARPs).
Keywords: Ataxia telangiectasia-mutated (ATM); DNA repair; Huntington’s disease; Oxidative stress; Poly ADP-ribose polymerase (PARP); Spinocerebellar ataxia.
Figures
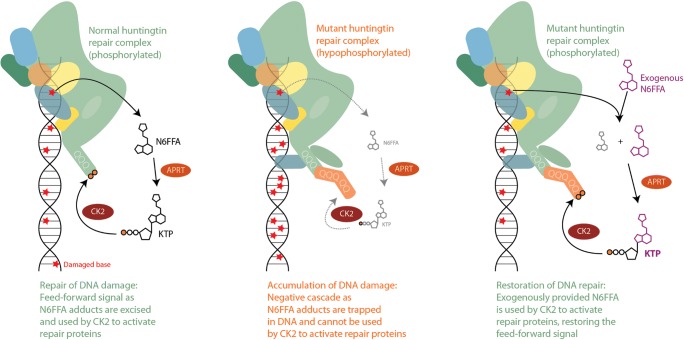
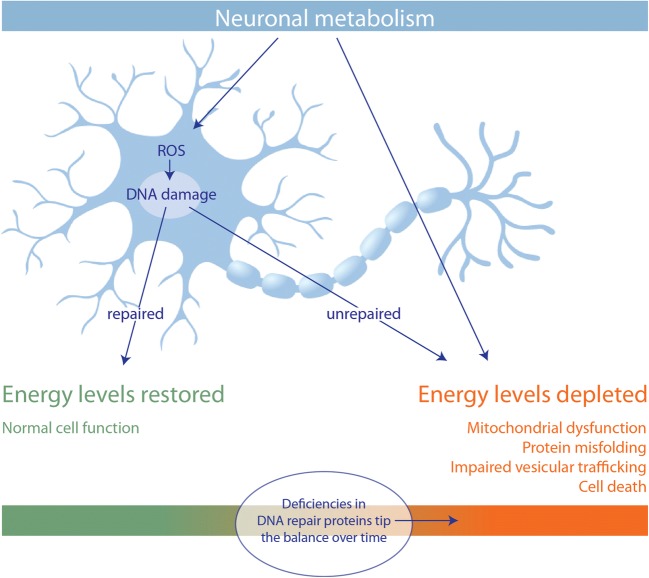
References
-
- Macdonald M. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. - PubMed
-
- Andrade MA, Bork P. HEAT repeats in the Huntington’s disease protein. Nat Genet. 1995;11:115–116. - PubMed
-
- Brandi V, Di Lella V, Marino M, Ascenzi P, Polticelli F. A comprehensive in silico analysis of huntingtin and its interactome. J Biomol Struct Dyn. 2018;36:3155–3171. - PubMed
-
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
