

Variation and selection on codon usage bias across an entire subphylum
- PMID: 31365533
- PMCID: PMC6701816
- DOI: 10.1371/journal.pgen.1008304
Variation and selection on codon usage bias across an entire subphylum
Erratum in
-
Correction: Variation and selection on codon usage bias across an entire subphylum.PLoS Genet. 2021 Sep 27;17(9):e1009824. doi: 10.1371/journal.pgen.1009824. eCollection 2021 Sep. PLoS Genet. 2021. PMID: 34570754 Free PMC article.
Abstract
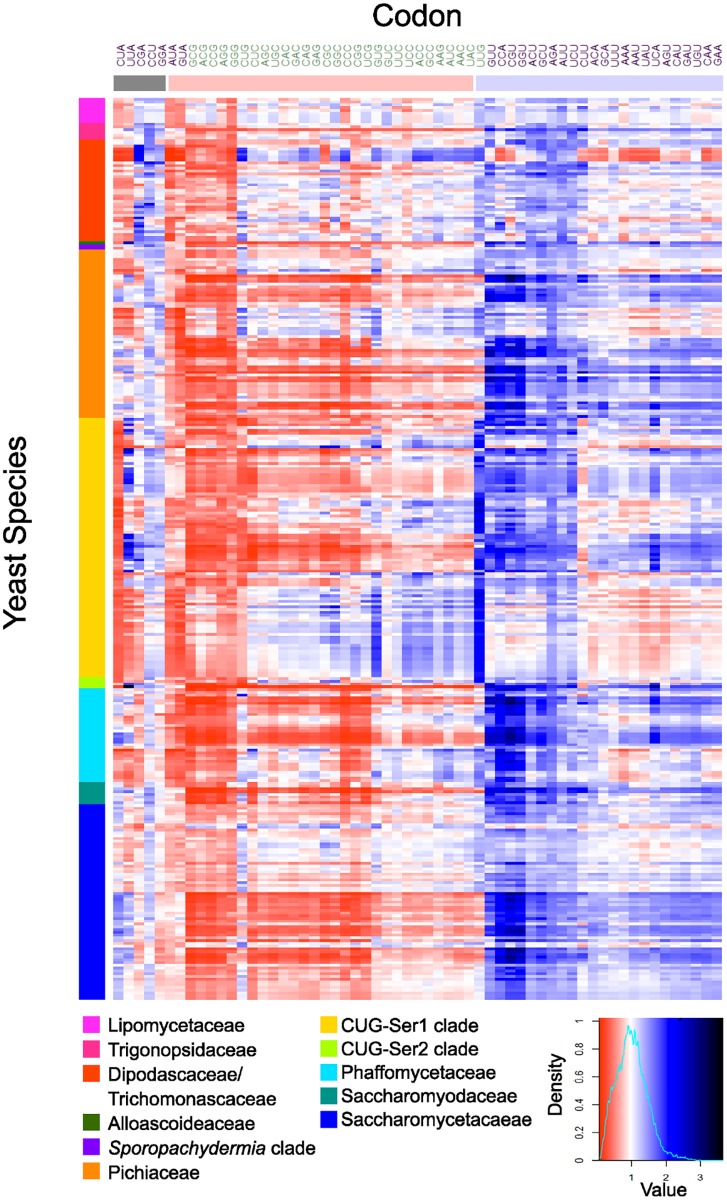
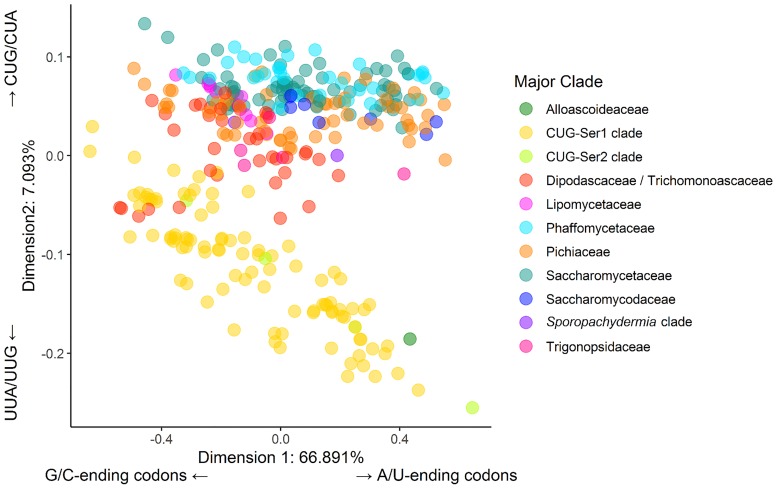
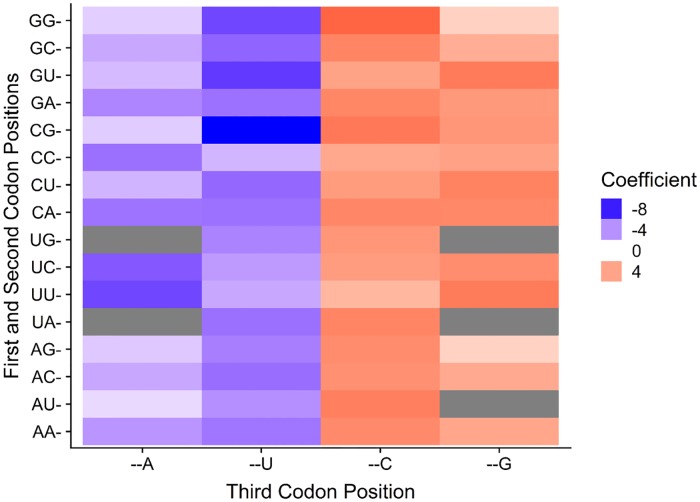
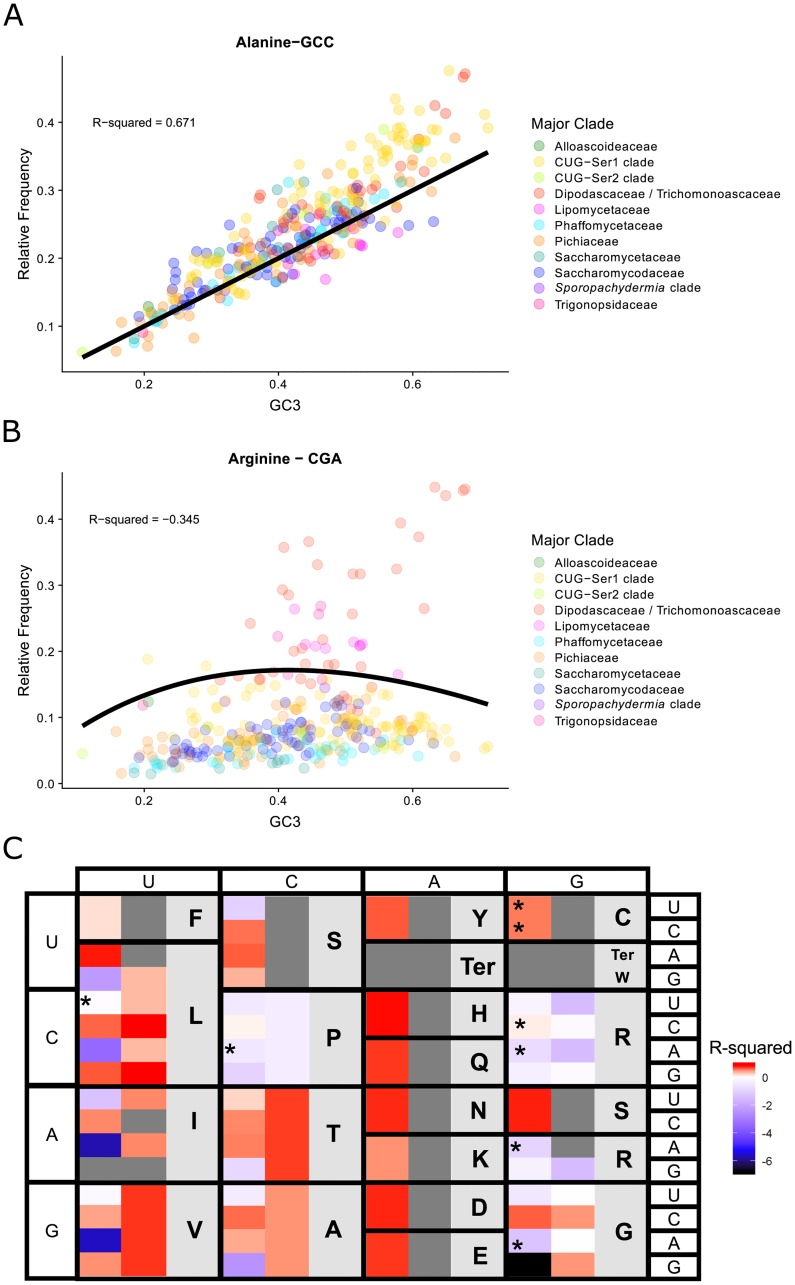
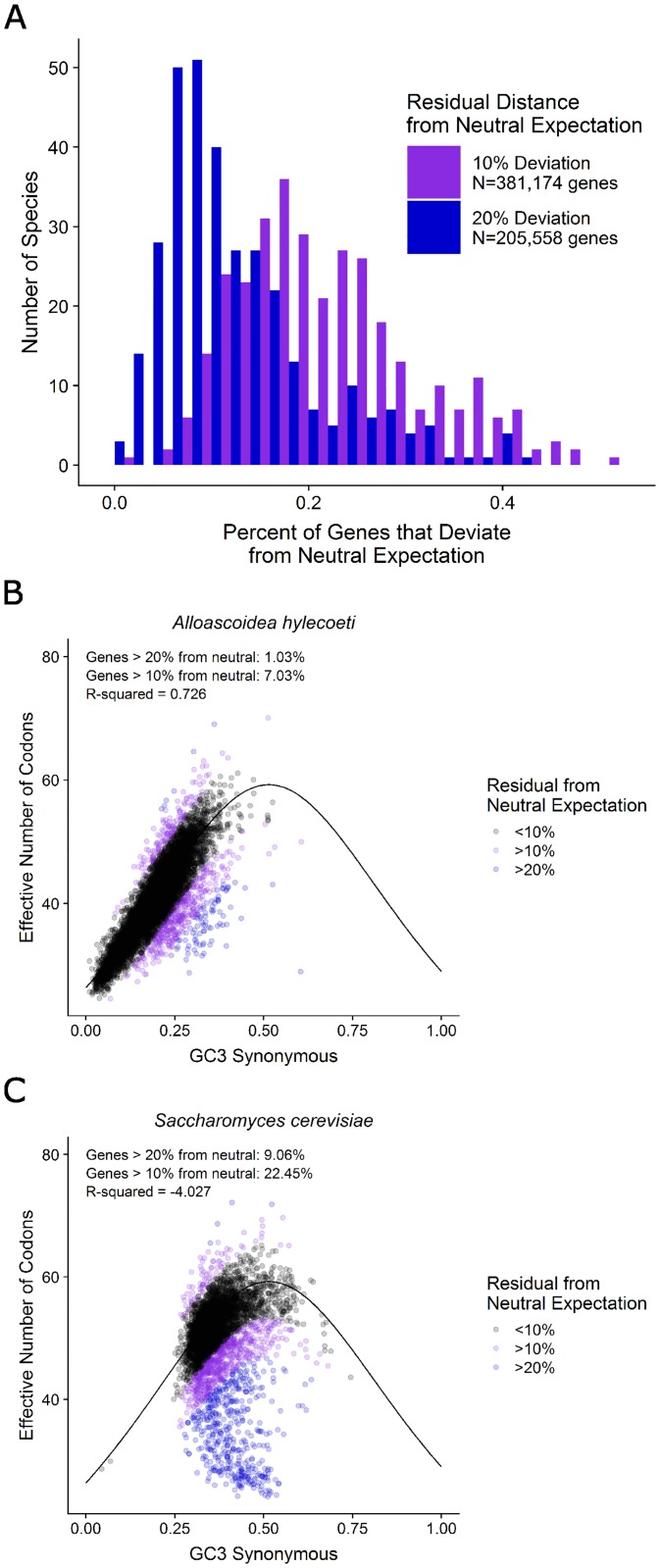
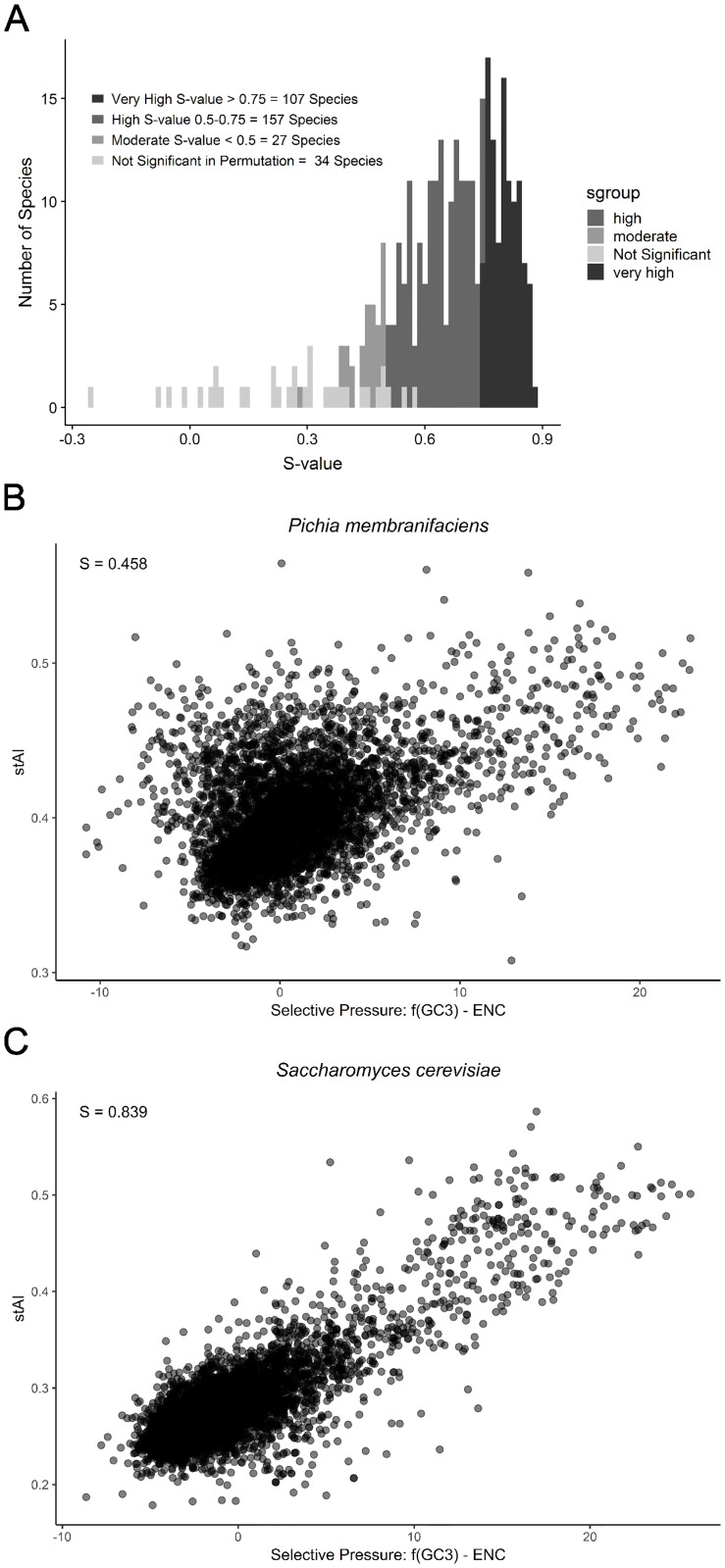
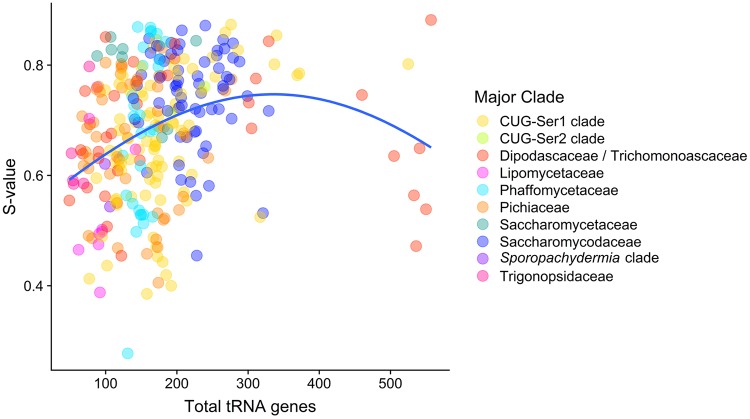
Variation in synonymous codon usage is abundant across multiple levels of organization: between codons of an amino acid, between genes in a genome, and between genomes of different species. It is now well understood that variation in synonymous codon usage is influenced by mutational bias coupled with both natural selection for translational efficiency and genetic drift, but how these processes shape patterns of codon usage bias across entire lineages remains unexplored. To address this question, we used a rich genomic data set of 327 species that covers nearly one third of the known biodiversity of the budding yeast subphylum Saccharomycotina. We found that, while genome-wide relative synonymous codon usage (RSCU) for all codons was highly correlated with the GC content of the third codon position (GC3), the usage of codons for the amino acids proline, arginine, and glycine was inconsistent with the neutral expectation where mutational bias coupled with genetic drift drive codon usage. Examination between genes' effective numbers of codons and their GC3 contents in individual genomes revealed that nearly a quarter of genes (381,174/1,683,203; 23%), as well as most genomes (308/327; 94%), significantly deviate from the neutral expectation. Finally, by evaluating the imprint of translational selection on codon usage, measured as the degree to which genes' adaptiveness to the tRNA pool were correlated with selective pressure, we show that translational selection is widespread in budding yeast genomes (264/327; 81%). These results suggest that the contribution of translational selection and drift to patterns of synonymous codon usage across budding yeasts varies across codons, genes, and genomes; whereas drift is the primary driver of global codon usage across the subphylum, the codon bias of large numbers of genes in the majority of genomes is influenced by translational selection.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Macroevolutionary changes in natural selection on codon usage reflect evolution of the tRNA pool across a budding yeast subphylum.Proc Natl Acad Sci U S A. 2025 Jul 8;122(27):e2419889122. doi: 10.1073/pnas.2419889122. Epub 2025 Jul 1. Proc Natl Acad Sci U S A. 2025. PMID: 40591602
-
Genomic factors shaping codon usage across the Saccharomycotina subphylum.G3 (Bethesda). 2024 Nov 6;14(11):jkae207. doi: 10.1093/g3journal/jkae207. G3 (Bethesda). 2024. PMID: 39213398 Free PMC article.
-
Genomic factors shaping codon usage across the Saccharomycotina subphylum.bioRxiv [Preprint]. 2024 May 24:2024.05.23.595506. doi: 10.1101/2024.05.23.595506. bioRxiv. 2024. Update in: G3 (Bethesda). 2024 Nov 6;14(11):jkae207. doi: 10.1093/g3journal/jkae207. PMID: 38826271 Free PMC article. Updated. Preprint.
-
Codon usage bias.Mol Biol Rep. 2022 Jan;49(1):539-565. doi: 10.1007/s11033-021-06749-4. Epub 2021 Nov 25. Mol Biol Rep. 2022. PMID: 34822069 Free PMC article. Review.
-
Codon Usage Bias: An Endless Tale.J Mol Evol. 2021 Dec;89(9-10):589-593. doi: 10.1007/s00239-021-10027-z. Epub 2021 Aug 12. J Mol Evol. 2021. PMID: 34383106 Review.
Cited by
-
Codon Usage Bias in Autophagy-Related Gene 13 in Eukaryotes: Uncovering the Genetic Divergence by the Interplay Between Nucleotides and Codon Usages.Front Cell Infect Microbiol. 2021 Nov 5;11:771010. doi: 10.3389/fcimb.2021.771010. eCollection 2021. Front Cell Infect Microbiol. 2021. PMID: 34804999 Free PMC article.
-
Genome-Wide Identification and Characterization of the Cystatin Gene Family in Bread Wheat (Triticum aestivum L.).Int J Mol Sci. 2021 Sep 23;22(19):10264. doi: 10.3390/ijms221910264. Int J Mol Sci. 2021. PMID: 34638605 Free PMC article.
-
Complete mitogenome characteristics and phylogenetic analysis of traditional Chinese medicinal plant Tinospora sagittata (Oliv.) Gagnep. from the Menispermaceae family.BMC Plant Biol. 2025 Feb 7;25(1):165. doi: 10.1186/s12870-025-06151-3. BMC Plant Biol. 2025. PMID: 39920585 Free PMC article.
-
Mitogenomics and mitochondrial gene phylogeny decipher the evolution of Saccharomycotina yeasts.Genome Biol Evol. 2022 May 3;14(5):evac073. doi: 10.1093/gbe/evac073. Genome Biol Evol. 2022. PMID: 35576568 Free PMC article.
-
Macroevolutionary changes in natural selection on codon usage reflect evolution of the tRNA pool across a budding yeast subphylum.Proc Natl Acad Sci U S A. 2025 Jul 8;122(27):e2419889122. doi: 10.1073/pnas.2419889122. Epub 2025 Jul 1. Proc Natl Acad Sci U S A. 2025. PMID: 40591602
References
-
- Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J Mol Biol. 1981;151(3):389–409. 10.1016/0022-2836(81)90003-6 . - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
