

Metabolic Dysregulations and Epigenetics: A Bidirectional Interplay that Drives Tumor Progression
- PMID: 31366176
- PMCID: PMC6721562
- DOI: 10.3390/cells8080798
Metabolic Dysregulations and Epigenetics: A Bidirectional Interplay that Drives Tumor Progression
Abstract
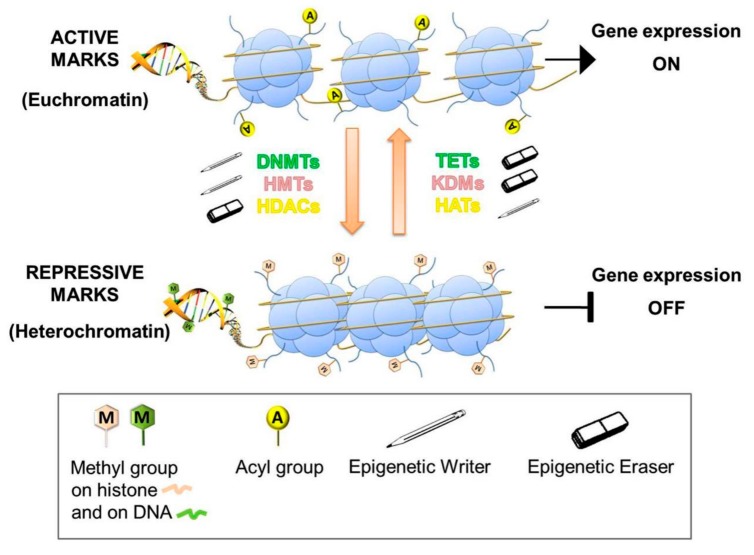
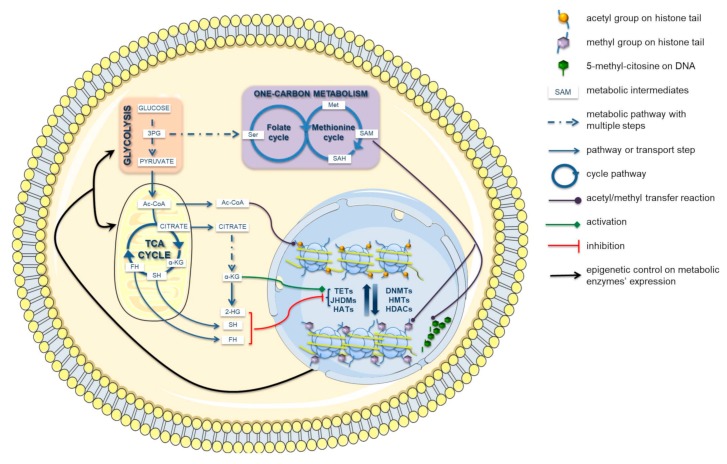
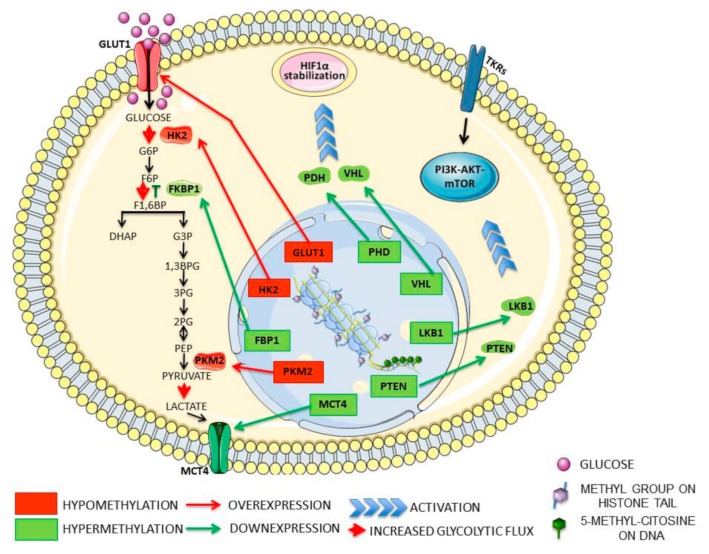
Cancer has been considered, for a long time, a genetic disease where mutations in keyregulatory genes drive tumor initiation, growth, metastasis, and drug resistance. Instead, theadvent of high-throughput technologies has revolutionized cancer research, allowing to investigatemolecular alterations at multiple levels, including genome, epigenome, transcriptome, proteome,and metabolome and showing the multifaceted aspects of this disease. The multi-omics approachesrevealed an intricate molecular landscape where different cellular functions are interconnected andcooperatively contribute to shaping the malignant phenotype. Recent evidence has brought to lighthow metabolism and epigenetics are highly intertwined, and their aberrant crosstalk can contributeto tumorigenesis. The oncogene-driven metabolic plasticity of tumor cells supports the energeticand anabolic demands of proliferative tumor programs and secondary can alter the epigeneticlandscape via modulating the production and/or the activity of epigenetic metabolites. Conversely,epigenetic mechanisms can regulate the expression of metabolic genes, thereby altering themetabolome, eliciting adaptive responses to rapidly changing environmental conditions, andsustaining malignant cell survival and progression in hostile niches. Thus, cancer cells takeadvantage of the epigenetics-metabolism crosstalk to acquire aggressive traits, promote cellproliferation, metastasis, and pluripotency, and shape tumor microenvironment. Understandingthis bidirectional relationship is crucial to identify potential novel molecular targets for theimplementation of robust anti-cancer therapeutic strategies.
Keywords: cancer; crosstalk; epigenetics; metabolism.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Cantor J.R., Sabatini D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012;2:881–898. doi: 10.1158/2159-8290.CD-12-0345. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
