

Plasmodium Genomics and Genetics: New Insights into Malaria Pathogenesis, Drug Resistance, Epidemiology, and Evolution
- PMID: 31366610
- PMCID: PMC6750138
- DOI: 10.1128/CMR.00019-19
Plasmodium Genomics and Genetics: New Insights into Malaria Pathogenesis, Drug Resistance, Epidemiology, and Evolution
Abstract
Protozoan Plasmodium parasites are the causative agents of malaria, a deadly disease that continues to afflict hundreds of millions of people every year. Infections with malaria parasites can be asymptomatic, with mild or severe symptoms, or fatal, depending on many factors such as parasite virulence and host immune status. Malaria can be treated with various drugs, with artemisinin-based combination therapies (ACTs) being the first-line choice. Recent advances in genetics and genomics of malaria parasites have contributed greatly to our understanding of parasite population dynamics, transmission, drug responses, and pathogenesis. However, knowledge gaps in parasite biology and host-parasite interactions still remain. Parasites resistant to multiple antimalarial drugs have emerged, while advanced clinical trials have shown partial efficacy for one available vaccine. Here we discuss genetic and genomic studies of Plasmodium biology, host-parasite interactions, population structures, mosquito infectivity, antigenic variation, and targets for treatment and immunization. Knowledge from these studies will advance our understanding of malaria pathogenesis, epidemiology, and evolution and will support work to discover and develop new medicines and vaccines.
Keywords: association studies; evolutionary selection; genetic mapping; genome diversity; population structure.
Copyright © 2019 American Society for Microbiology.
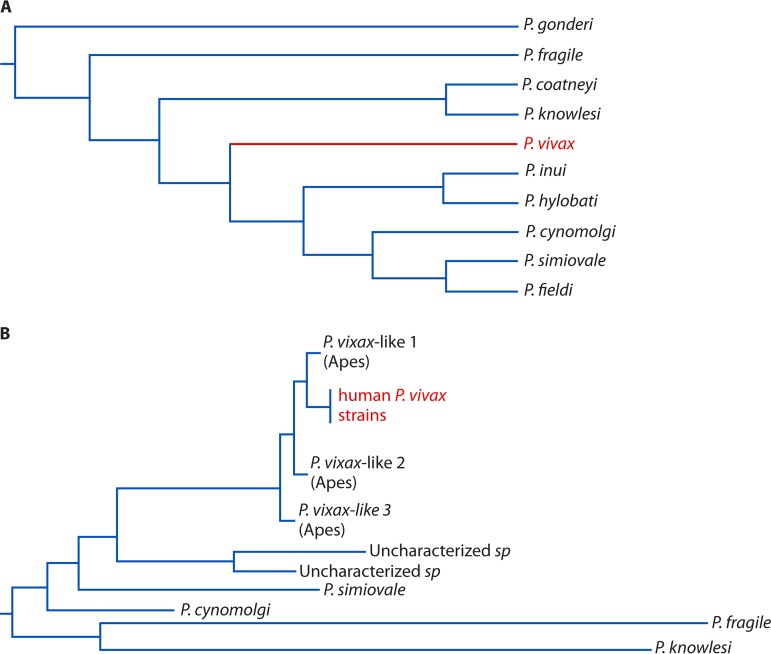
Figures
References
-
- Noor AM, Kinyoki DK, Mundia CW, Kabaria CW, Mutua JW, Alegana VA, Fall IS, Snow RW. 2014. The changing risk of Plasmodium falciparum malaria infection in Africa: 2000-10: a spatial and temporal analysis of transmission intensity. Lancet 383:1739–1747. doi: 10.1016/S0140-6736(13)62566-0. - DOI - PMC - PubMed
-
- Carlton JM, Angiuoli SV, Suh BB, Kooij TW, Pertea M, Silva JC, Ermolaeva MD, Allen JE, Selengut JD, Koo HL, Peterson JD, Pop M, Kosack DS, Shumway MF, Bidwell SL, Shallom SJ, van Aken SE, Riedmuller SB, Feldblyum TV, Cho JK, Quackenbush J, Sedegah M, Shoaibi A, Cummings LM, Florens L, Yates JR, Raine JD, Sinden RE, Harris MA, Cunningham DA, Preiser PR, Bergman LW, Vaidya AB, van Lin LH, Janse CJ, Waters AP, Smith HO, White OR, Salzberg SL, Venter JC, Fraser CM, Hoffman SL, Gardner MJ, Carucci DJ. 2002. Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature 419:512–519. doi: 10.1038/nature01099. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
