

A comprehensive examination of Nanopore native RNA sequencing for characterization of complex transcriptomes
- PMID: 31366910
- PMCID: PMC6668388
- DOI: 10.1038/s41467-019-11272-z
A comprehensive examination of Nanopore native RNA sequencing for characterization of complex transcriptomes
Abstract
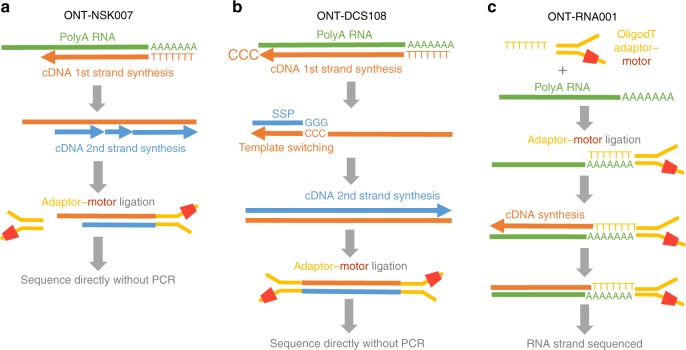
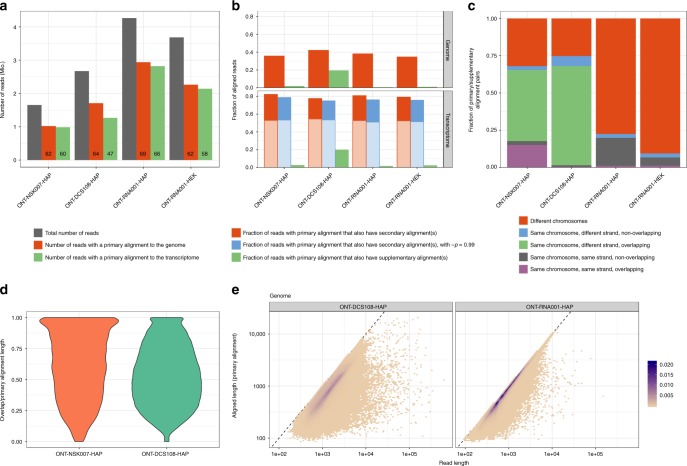
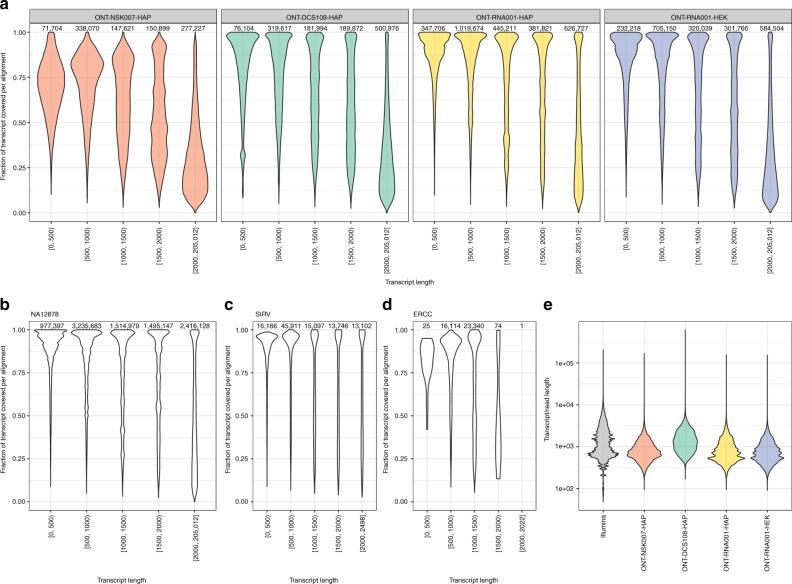
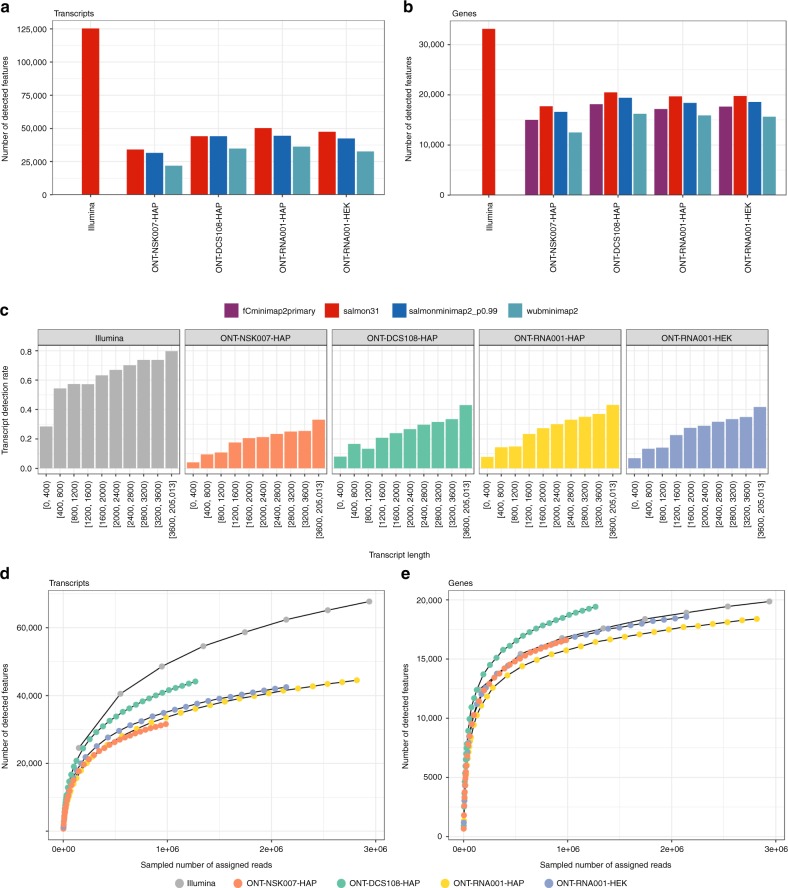
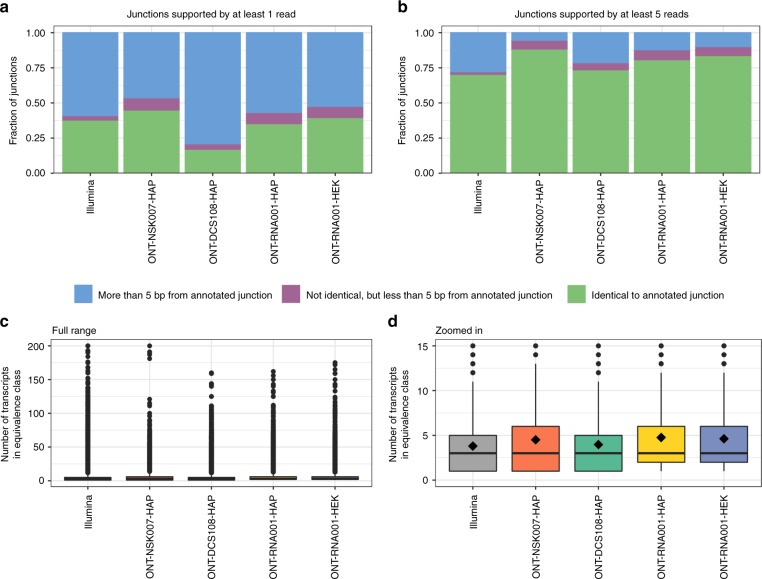
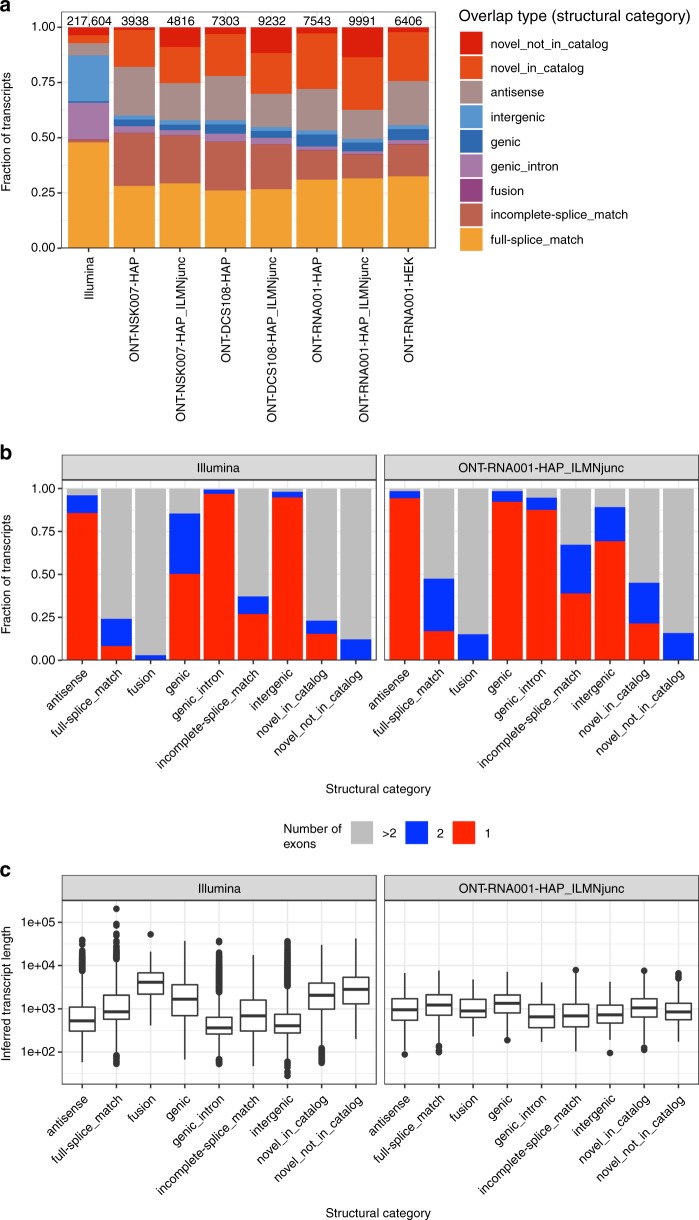
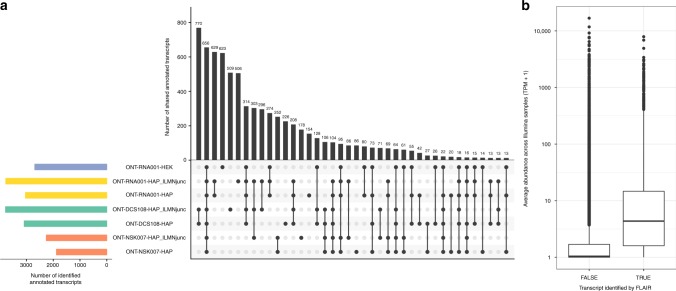
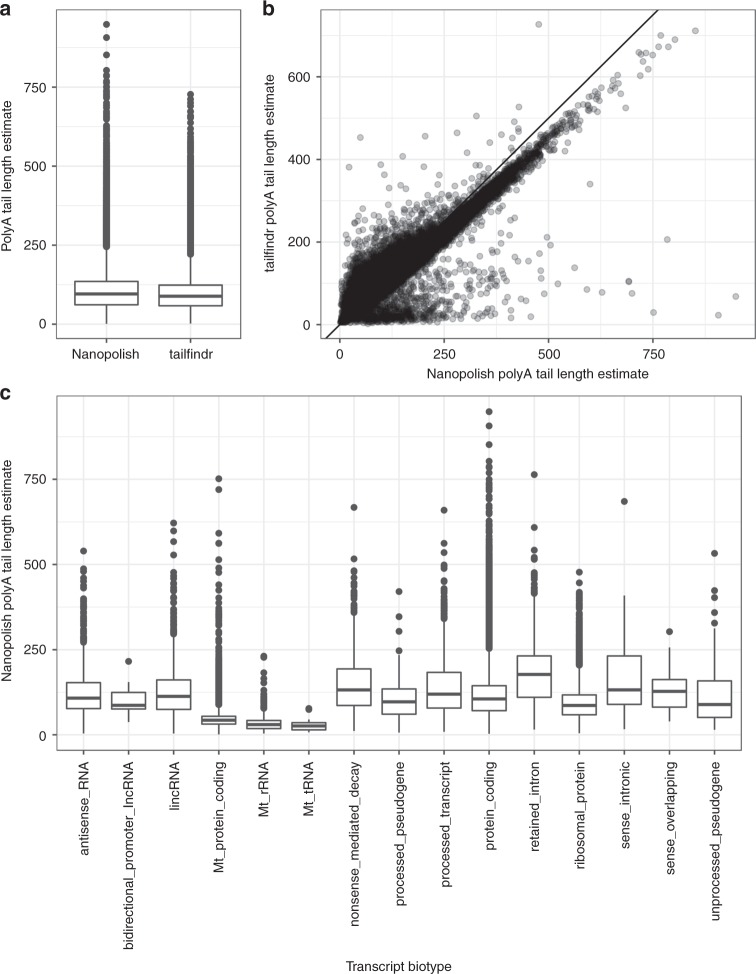
A platform for highly parallel direct sequencing of native RNA strands was recently described by Oxford Nanopore Technologies, but despite initial efforts it remains crucial to further investigate the technology for quantification of complex transcriptomes. Here we undertake native RNA sequencing of polyA + RNA from two human cell lines, analysing ~5.2 million aligned native RNA reads. To enable informative comparisons, we also perform relevant ONT direct cDNA- and Illumina-sequencing. We find that while native RNA sequencing does enable some of the anticipated advantages, key unexpected aspects currently hamper its performance, most notably the quite frequent inability to obtain full-length transcripts from single reads, as well as difficulties to unambiguously infer their true transcript of origin. While characterising issues that need to be addressed when investigating more complex transcriptomes, our study highlights that with some defined improvements, native RNA sequencing could be an important addition to the mammalian transcriptomics toolbox.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
